
Sr. Director:
Lenalidomida es un fármaco relacionado con talidomida, con el que comparte sus propiedades inmunomoduladoras, antiangiogénicas y antineoplásicas, designado por la European Medicine Agency (EMEA) como medicamento huérfano en 2001. En combinación con dexametasona está indicada (FDA y EMEA) en el tratamiento del mieloma múltiple (MM) en los pacientes que hayan recibido un tratamiento previo1. También está indicada (FDA) para el tratamiento de pacientes con anemia dependiente de transfusiones, producida por síndromes mielodisplásicos de riesgo bajo o intermedio asociados a la anomalía citogenética 5q, con o sin anomalías citogenéticas adicionales, indicación que fue denegada por la EMEA en 20072.
No está exenta de efectos adversos entre los que destacan: toxicidad hematológica (trombocitopenia y neutropenia), neuropatías, accidentes tromboembólicos, diarrea, exantemas, fatiga, infecciones y síntomas osteomusculares. Ante el riesgo de teratogenicidad, para evitar la exposición fetal al medicamento, se encuentra bajo un programa de control de riesgos específico. Recientemente ha sido comercializada en España, para MM, y con la categoría de diagnóstico hospitalario1.
Tanto por la novedad terapéutica que supone, como por la frecuencia y severidad de los efectos adversos, consideramos interesante comunicar nuestra experiencia con este fármaco en 15 pacientes tratados en nuestro hospital, previa a su comercialización y bajo tratamiento compasivo, centrándonos en los aspectos de seguridad que motivaron modificaciones en las pautas previstas, como base para una reflexión a propósito de su comercialización.
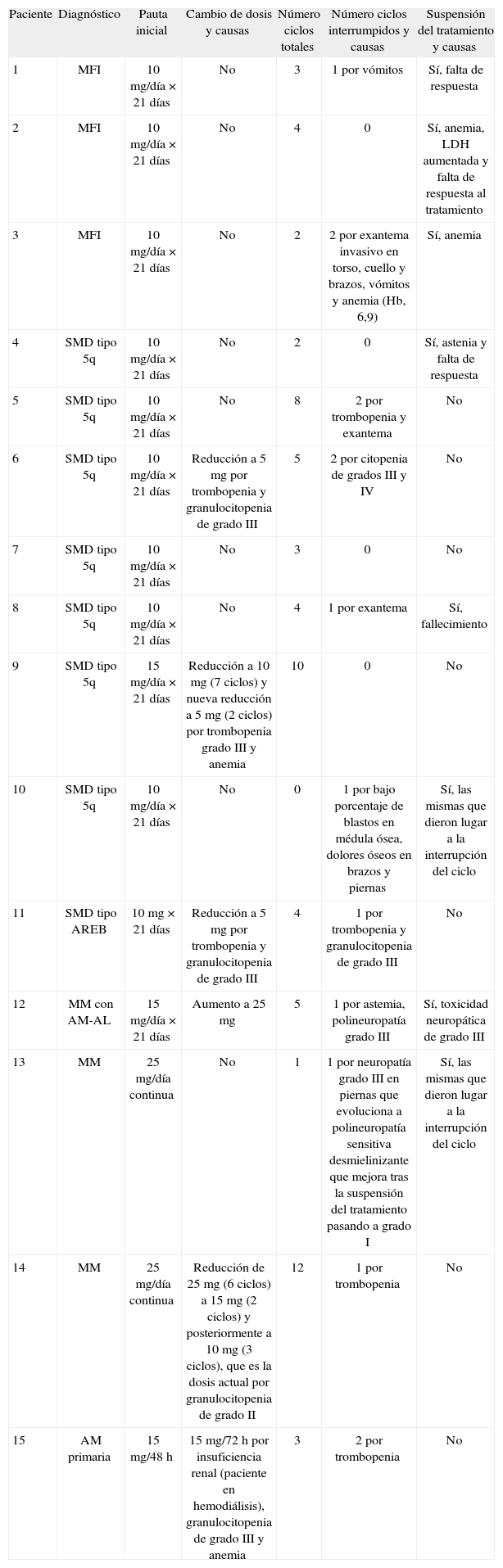
Descripción de la serie de casosDe los 15 pacientes tratados, 5 son mujeres (33,3%) y 10 varones (66,6%). Todos comenzaron con la dosis estándar para su proceso: 25 mg/día para MM3, 10 mg/día para el síndrome mielodisplásico (SMD)4, 10 mg/día para mielofibrosis idiomática5 y 15 mg/día en amiloidosis primaria6 (tabla 1).
Pacientes tratados con lenalidomida: diagnóstico, pautas y modificaciones
| Paciente | Diagnóstico | Pauta inicial | Cambio de dosis y causas | Número ciclos totales | Número ciclos interrumpidos y causas | Suspensión del tratamiento y causas |
| 1 | MFI | 10 mg/día × 21 días | No | 3 | 1 por vómitos | Sí, falta de respuesta |
| 2 | MFI | 10 mg/día × 21 días | No | 4 | 0 | Sí, anemia, LDH aumentada y falta de respuesta al tratamiento |
| 3 | MFI | 10 mg/día × 21 días | No | 2 | 2 por exantema invasivo en torso, cuello y brazos, vómitos y anemia (Hb, 6,9) | Sí, anemia |
| 4 | SMD tipo 5q | 10 mg/día × 21 días | No | 2 | 0 | Sí, astenia y falta de respuesta |
| 5 | SMD tipo 5q | 10 mg/día × 21 días | No | 8 | 2 por trombopenia y exantema | No |
| 6 | SMD tipo 5q | 10 mg/día × 21 días | Reducción a 5 mg por trombopenia y granulocitopenia de grado III | 5 | 2 por citopenia de grados III y IV | No |
| 7 | SMD tipo 5q | 10 mg/día × 21 días | No | 3 | 0 | No |
| 8 | SMD tipo 5q | 10 mg/día × 21 días | No | 4 | 1 por exantema | Sí, fallecimiento |
| 9 | SMD tipo 5q | 15 mg/día × 21 días | Reducción a 10 mg (7 ciclos) y nueva reducción a 5 mg (2 ciclos) por trombopenia grado III y anemia | 10 | 0 | No |
| 10 | SMD tipo 5q | 10 mg/día × 21 días | No | 0 | 1 por bajo porcentaje de blastos en médula ósea, dolores óseos en brazos y piernas | Sí, las mismas que dieron lugar a la interrupción del ciclo |
| 11 | SMD tipo AREB | 10 mg × 21 días | Reducción a 5 mg por trombopenia y granulocitopenia de grado III | 4 | 1 por trombopenia y granulocitopenia de grado III | No |
| 12 | MM con AM-AL | 15 mg/día × 21 días | Aumento a 25 mg | 5 | 1 por astemia, polineuropatía grado III | Sí, toxicidad neuropática de grado III |
| 13 | MM | 25 mg/día continua | No | 1 | 1 por neuropatía grado III en piernas que evoluciona a polineuropatía sensitiva desmielinizante que mejora tras la suspensión del tratamiento pasando a grado I | Sí, las mismas que dieron lugar a la interrupción del ciclo |
| 14 | MM | 25 mg/día continua | Reducción de 25 mg (6 ciclos) a 15 mg (2 ciclos) y posteriormente a 10 mg (3 ciclos), que es la dosis actual por granulocitopenia de grado II | 12 | 1 por trombopenia | No |
| 15 | AM primaria | 15 mg/48 h | 15 mg/72 h por insuficiencia renal (paciente en hemodiálisis), granulocitopenia de grado III y anemia | 3 | 2 por trombopenia | No |
AM: amiloidosis; AREB: anemia refractaria con exceso de blastos; LDH: lactato deshidrogenasa; MFI: mielofibrosis idiomática; MM: mieloma múltiple; SMD: síndrome mielodisplásico.
Sólo en 2 (ambos con SMD 5q) de los 15 pacientes se pudo administrar la dosis y pauta programada, y fue eficaz y bien tolerada. En los 13 restantes (86,6%) fue necesario modificar el esquema terapéutico inicialmente previsto como consecuencia de los efectos adversos hematológicos y neurológicos que presentaron.
En 11 pacientes (73,3%) se optó por suspender temporalmente la administración del fármaco, reiniciando el tratamiento una vez recuperada una situación clínica aceptable.
Por otra parte, el 86,6% de los pacientes precisó tratamiento concomitante con filgrastrim durante una mediana de 4 días.
Independientemente de la enfermedad subyacente, los efectos adversos más frecuentes fueron astenia moderada y alteraciones en el sueño, presentes en todos los pacientes al comienzo del tratamiento y que en 5 de ellos (33,3%) remitieron.
Por patologías, en SMD, 3 pacientes (37,5%) manifestaron neutropenias de grados II-III (500-1.500 neutrófilos/ml), en 4 (50%) hubo trombopenia con cifras de 50.000-75.000 plaquetas/ml; 3 (37,5%) presentaron exantemas, y 1 (12,5%) refirió dolores óseos.
Los mismos efectos adversos aparecieron en los pacientes con MM, excepto por una menor mielotoxicidad.
Las reacciones adversas y su incidencia son similares a las descritas en la bibliografía2,6-8 con la excepción de las polineuropatías en los pacientes con MM que fueron más frecuentes en nuestra serie (ocurriendo en 2 de los 3 pacientes). Estos efectos adversos fueron notificados al Sistema Español de Farmacovigilancia.
DiscusiónCon las limitaciones propias de una pequeña y heterogénea serie de casos, pero con la ventaja de mostrar la realidad asistencial fuera del contexto sobreprotegido de los ensayos clínicos, lenalidomida se presenta en nuestro estudio como una herramienta terapéutica potente con la que se requiere un seguimiento clínico minucioso que debe ir más allá del programa de gestión de riesgos específico. Muy por delante de la falta de respuesta, los efectos adversos observados, variados y severos, son el elemento limitante del tratamiento.
En nuestra serie todos los pacientes recibieron en fármaco con protocolo de uso compasivo, y al realizarse la dispensación en el servicio de farmacia los pacientes se han beneficiado de una atención farmacéutica especializada y de una fluida comunicación con el equipo de hematólogos que atienden estos procesos. Esta atención especial hace que de modo no reglado se realice una farmacovigilancia activa, que no sólo facilita la toma de decisiones precozmente, evitando problemas relacionados con la medicación, sino que además permite estudios como el presente, en el que se registra y difunde información de seguridad, contribuyendo a aumentar nuestros conocimientos acerca de un fármaco que si bien se perfila como una alternativa eficiente, también presenta contrapartidas en materia de seguridad.
En estas condiciones, creemos que la comercialización del fármaco con la consideración de diagnóstico hospitalario no parece aportar ninguna ventaja frente a la alternativa de uso hospitalario, y así parecen haberlo entendido algunos sistemas de salud de determinadas comunidades autónomas, que han limitado su dispensación al ámbito hospitalario. Al evitar la centralización de la dispensación en los servicios de farmacia se pierde, o al menos se dificulta, la continuidad en los cuidados del paciente; el seguimiento terapéutico y la atención farmacéutica continuada pueden verse perjudicados, y pensamos que con ello no se contribuye a un uso más seguro de un fármaco novedoso, prometedor pero complejo. El seguimiento analítico y clínico que procede con estos procesos exige visitas frecuentes al hospital, por lo que la dispensación en el servicio de farmacia no debería suponer un esfuerzo gravoso para el paciente. En cualquier caso, la comodidad no parece un argumento relevante en un escenario en el que casi el 90% de los pacientes ven modificadas sus pautas como consecuencia de los efectos adversos.
A todo ello, la dispensación fuera del hospital suma otra notable desventaja para el sistema sanitario, no desdeñable considerando el precio del fármaco que, dependiendo de la dosis, ronda los 6.000 € por envase. Tanto las reducciones de dosis, como las interrupciones de los ciclos, conllevan un desaprovechamiento del fármaco, que en nuestro caso podría alcanzar un coste potencial muy importante. Manteniendo la dispensación dentro del ámbito hospitalario, el seguimiento farmacéutico permite, como valor añadido a la labor de farmacovigilancia, una optimización de la gestión económica.
Posiblemente en el establecimiento de la categoría de diagnóstico hospitalario para lenalidomida no se hayan valorado suficientemente los argumentos que aquí presentamos.

