
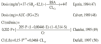


REVISIONES
ESTADO ACTUAL DE LA INDIVIDUALIZACIÓN POSOLÓGICA EN QUIMIOTERAPIA ANTINEOPLÁSICA
Jiménez Torres, N. V., Doctor en Farmacia, Jefe de Servicio de Farmacia, Catedrático del Departamento de Farmacia y Tecnología Farmacéutica; Pérez Ruixo, J. J., Licenciado en Farmacia, Residente III año; Medina Hernández, C. A., Licenciado en Farmacia, Becaria de AFAHPE; Llopis García, M. C., Licenciado en Farmacia, Becaria FIS
Servicio de Farmacia. Hospital Universitario Dr. Peset. Valencia. Departamento de Farmacia y Tecnología Farmacéutica. Universidad de Valencia
Correspondencia: N. V. Jiménez Torres. Hospital Dr. Peset. Servicio de Farmacia. Avda. Gaspar Aguilar, 90. 46017 Valencia. E-mail: nvictor@san.gva.es
Fecha de recepción: 30-11-1998
Palabras clave:
Quimioterapia antineoplásica. Farmacocinética. Farmacodinamia.
Resumen:
Una característica principal del tratamiento farmacológico antineoplásico es la variabilidad en la respuesta clínica deseada. El interés por conocer y cuantificar las distintas fuentes de variabilidad en la respuesta farmacológica se fundamenta en la hipótesis de que reducir variabilidad en la disposición por el organismo a los antineoplásicos es disminuir variabilidad en su respuesta clínica, tanto en términos de eficacia como de toxicidad y calidad de vida de los pacientes. En base a lo que antecede este trabajo revisa, con criterios farmacocinéticos y farmacodinámicos actuales, los factores determinantes de la individualización posológica en quimioterapia antineoplásica. Asimismo analiza los métodos que soportan tal criterio posológico y su validación.
CURRENT STATUS OF INDIVIDUALIZED DOSING OF ANTINEOPLASTIC CHEMOTHERAPY
Key words:
Antineoplastic chemotherapy. Pharmacokinetics. Pharmacodynamics.
Summary:
One of the main characteristics of anticancer pharmacology is variable clinical response. Identifying and quantifying sources of variability in the pharmacological response is interesting because if individual susceptibility to anticancer agents can be determined, the variability of clinical response in terms of effectiveness, toxicity and impact on quality of life can of reduced. A review was made of current pharmacokinetic and pharmacodynamic criteria, the determinant factors of individual dosing in antineoplastic chemotherapy. Methods for determining dosage and their validation are analyzed.
INTRODUCCIÓN
La utilización de los principios y métodos farmacocinéticos y farmacodinámicos para individualizar la posología en pacientes oncológicos es una práctica cada día más introducida en clínica. Esta metodología, debido al bajo índice terapéutico de los medicamentos antineoplásicos, trata de optimizar la respuesta en el paciente, considerado individualmente, desarrollando y promoviendo estrategias farmacológicas que garanticen la utilización racional de estos medicamentos (1).
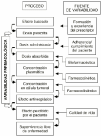
Una característica principal del tratamiento farmacológico antineoplásico es la variabilidad en la respuesta clínica deseada. Las razones o determinantes de esta variabilidad se han tratado de fundamentar en los complejos procesos intracelulares y en los mecanismos de acción y de resistencia de los citostáticos, por lo que la potencial relación de la dosis con la respuesta todavía se sigue utilizando como criterio para predecir la respuesta en el paciente oncológico (2, 3). Sin embargo, el concepto probabilístico de dosis-respuesta se encuentra prácticamente superado en base a que la variabilidad en los procesos que acontecen, desde la prescripción del tratamiento hasta la respuesta en el paciente, es multifactorial y no sólo explicable por la dosis (Fig. 1).
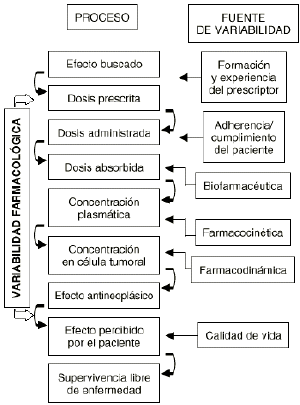
Figura 1.--Procesos y fuentes de variabilidad en la respuesta farmacológica.
El interés por conocer y cuantificar las distintas fuentes de variabilidad en la respuesta farmacológica se fundamenta en la hipótesis de que reducir variabilidad en la disposición por el organismo a los antineoplásicos es disminuir variabilidad en su respuesta clínica, tanto en términos de eficacia como de toxicidad y calidad de vida de los pacientes (2). Este objetivo se alcanza de forma más directa cuando se estudia la relación existente entre la concentración de fármaco en el organismo y su respuesta clínica que cuando se hace con respecto a la dosis respuesta, ya que para definir las bases científicas de la farmacoterapia generalmente se requiere hacer referencia a la evolución temporal de sus concentraciones plasmáticas (4, 5).
La consecución de resultados óptimos en pacientes diagnosticados de cáncer presenta, además de la variabilidad antes referida, una barrera adicional derivada del estrecho índice terapéutico que presentan la mayoría de los fármacos antineoplásicos. Esta circunstancia supone una dificultad añadida para definir la dosis óptima que, no obstante, siempre implica un riesgo de toxicidad en el paciente oncológico superior al inherente en el tratamiento farmacoterapéutico de otras enfermedades.
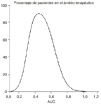
La representación gráfica del porcentaje de pacientes cuyo tratamiento ha sido efectivo y ausente de toxicidad y su correspondiente valor de área bajo la curva concentración plasmática-tiempo (AUC), es una curva de Gauss (Fig. 2) que expresa el ámbito terapéutico de los fármacos de modo que cuanto menor sea su máximo y amplitud, más estrecho será el mismo. En consecuencia, uno de los principales objetivos de la oncología moderna es ampliar el margen terapéutico de la quimioterapia antineoplásica. Este reto se ha abordado desde frentes tan diferentes como la utilización de combinaciones de fármacos con distinta toxicidad limitante de dosis, la quimioterapia de intensificación, el empleo de fármacos de «rescate» de los tejidos no tumorales como la administración de ácido folínico y/o factores estimulantes de colonias o bien prácticas terapéuticas como el trasplante de médula ósea o más recientemente la reinfusión de progenitores hematopoyéticos de sangre periférica. En este mismo escenario se sitúa la utilización de moduladores biológicos, como ciclosporina, cuyo objetivo es disminuir los mecanismos específicos de resistencia de las células tumorales, como es el caso de la glicoproteína P (6, 7).
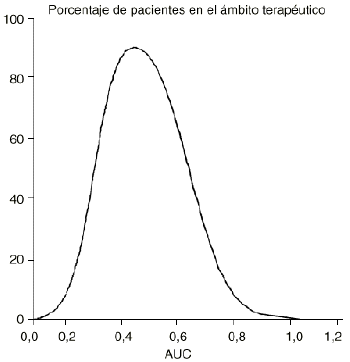
Figura 2.--Porcentaje de pacientes en el ámbito terapéutico en función del área bajo la curva concentración-tiempo (AUC).
La investigación en estas líneas de actuación y el creciente conocimiento sobre el proceso molecular subyacente en el crecimiento neoplásico son los pilares básicos para establecer científicamente las bases farmacoterapéuticas para el tratamiento del cáncer en un futuro no muy lejano.
FACTORES DETERMINANTES DEL AJUSTE DE DOSIS
La optimización de la dosis o individualización posológica en oncología, basada en la monitorización de la respuesta del citostático, exige definir criterios de eficacia como puede ser la concentración plasmática pic por ejemplo, metotrexato (8)--, el tiempo durante el cual las concentraciones se encuentran por encima de un determinado valor umbral --por ejemplo, etopósido (29)-- o, más frecuentemente, el AUC --por ejemplo, carboplatino (9)-- o el área sobre la curva de leucocitos-tiempo (AOC) --por ejemplo, epirrubicina (11)--. Estos criterios, a su vez, dependen de factores antropométricos, bioquímicos, farmacodinámicos y farmacocinéticos, o bien indicadores de funcionalidad orgánica, entre otros. El objetivo común de la individualización posológica o búsqueda de la dosis óptima individual es alcanzar y mantener la máxima eficacia anticancerígena, con una toxicidad aceptable para los pacientes, a fin de lograr la remisión de la enfermedad o el máximo tiempo libre de enfermedad.
La dosificación basada en características antropométricas, como el peso corporal total, peso magro o bien la superficie corporal, es función del peso y altura del paciente está arraigada en clínica por su tradición y por estimar que la medida del tamaño corporal informa del tamaño y función del órgano eliminador del fármaco; es decir, se establece una potencial relación entre el aclaramiento sistémico de los fármacos antineoplásicos y estos parámetros antropométricos. Sin embargo, la variabilidad interindividual del aclaramiento sistémico de la mayoría de los antineoplásicos, que pueden explicar estas covariables, es mínima comparada con la variabilidad interindividual inherente al propio fármaco. Por ejemplo, la utilización del peso corporal total en el modelo de predicción del aclaramiento del 5-fluoruracilo (5FU) justifica menos del 5% de su variabilidad (10). En esta línea de razonamiento se explicaría por qué en pacientes con cáncer colorrectal la dosis calculada de 5FU en base a la superficie corporal posee tanta variabilidad clínica como para originar en los pacientes desde una respuesta deseada, con ausencia de toxicidad, hasta enterocolitis de pronóstico fatal (2). En la tabla 1 se describen algunos de los agentes antineoplásicos más utilizados en la actualidad y su relación con la superficie corporal (3). Para algunos fármacos, como es el caso de la epirrubicina, la medida del peso magro, como indicador del tamaño corporal, se ha sugerido como mejor variable predictora que la superficie corporal, para su correlación con el aclaramiento sistémico (11).
| Tabla 1. Parámetros farmacocinéticos de antineoplásicos y superficie corporal | ||
| Fármaco | Observaciones | Referencias |
| Carboplatino | El Cl se correlaciona con la FG El Cl se correlaciona con la SC. | Madden, T (1992) (30) Newell, DR (1993) (40) |
| Epirrubicina | El Cl no se correla ciona con la SC. | Cosolo, WC (1994) (41) |
| Etopósido | El AUC y Cl no se correlaciona con la SC. | Lowis, SP (1994) (42) Ratain, MJ (1991) (43) |
| 5FU | Hay controversia. | Climente, M (1997) (10) |
| Ifosfamida | El Cl, Vd y t1/2 aumen tan con la SC. | Boddy, AV (1993) (44) |
| Paclitaxel | El Cl se correlaciona con la altura, pero no con SC. | Sonnichsen, DS (1994) (45) Grochow, LB (1990) (46) |
| Cl: aclaramiento plasmático. AUC: área bajo la curva. Vd: volumen de distribución. t1/2: semivida biológica. SC: superficie corporal. FG: filtración glomerular. Tomada de Gurney et al (3). | ||
Actualmente, pues, además de los parámetros antropométricos se buscan indicadores de funcionalidad de órganos vitales que correlacionen bien con los parámetros farmacocinéticos y/o farmacodinámicos y, en consecuencia, permitan optimizar la dosis.
Los parámetros fisiológicos, bioquímicos-hematológicos no se han estudiado aún con la misma intensidad que los antropométricos, pero se ha encontrado correlación en la eficacia y/o toxicidad de algunos citostáticos para enzimas hepáticos, creatinina plasmática, unión a proteínas plasmáticas, neutrófilos y leucocitos, entre otros (2). Ahora bien, cuando los criterios basados en estos parámetros se aplican a priori o empíricamente a la posología, si bien se evitan las reacciones adversas graves que podrían producirse en el paciente al administrar la dosis estándar, la efectividad del tratamiento se ve disminuida como consecuencia de la reducción de dosis que se administra. Por ejemplo, ante un paciente con alteración de la función renal o hepática es común, en oncología práctica, retrasar el tratamiento (ciclo) o reducir la dosis en un 25-50%. De hecho, las directrices posológicas existentes, excepto para fármacos muy concretos (carboplatino y docetaxel), resultan conflictivas (3).
De este grupo de indicadores el mejor estudiado es la función renal. No obstante, la individualización posológica del carboplatino, basada sólo en la tasa de filtración glomerular del paciente mejora su predicción al incorporar en el modelo, como variables independientes, otras características fisiopatológicas del paciente (tabla 2) (9). Por tanto, la inclusión de nuevas variables en el modelo se han de contemplar a fin de evaluar si mejora la exactitud y precisión inicial.
Tabla 2. Modelos desarrollados para la individualización posológica de carboplatino a priori | |
| Modelo | Referencia |

| |
| *%Rp: porcentaje de reducción de plaquetas deseado. BSA: superficie corporal, expresada en m2. Clcr: aclaramiento de creatinina, expresado en ml/min. AUC: área bajo la curva, expresada en mg/l·h. P: peso expresado en kg. E: edad, expresada en años. S: sexo, toma el valor de 1 cuando es mujer. FG: filtración glomerular, expresada en ml/min. | |
Otros citostáticos, como por ejemplo irinotecan y docetaxel, claramente se relacionan con más de un tipo de variables antropométricas, entre ellas el peso corporal. Así, el aclaramiento de docetaxel es predicho según la siguiente ecuación (13):
Cl(L/h) = BSA · [22,1 (3,55 · AAG) (0,095 · edad) + (0,225 · Alb)] · (1 0,334 · Hep)
donde BSA es la superficie corporal, expresada en m2; AAG es la *1-glicoproteína ácida, expresada en UI/l; la edad se expresa en años; Alb es la albúmina plasmática, expresada en g/l, y Hep es una variable dicotómica que cuantifica la presencia de insuficiencia hepática, de tal forma que toma el valor de 1 cuando el paciente presenta ciertos enzimas hepáticos elevados y 0 cuando esto no ocurre. La utilización de estas covariables en el ajuste de dosis permite reducir la variabilidad interindividual del 50 al 33% (14). Este modelo evidencia a su vez cómo los parámetros de funcionalidad hepática tienen mayor capacidad de predicción del aclaramiento hepático del fármaco que los parámetros antropométricos.
En suma, la incorporación de parámetros bioquímicos-hematológicos y/o de indicadores de la funcionalidad hepática o renal, además de los antropométricos, a los modelos de dosificación individualizada de determinados antineoplásicos, reduce la variabilidad del AUC y/o del aclaramiento del fármaco. Ahora bien, la consideración de sólo estos criterios a la hora de prescribir un agente citostático no representa una verdadera individualización posológica debido a la amplia variabilidad interindividual inexplicable y a la dificultad de su discriminación y cuantificación.
Los parámetros farmacocinéticos más habitualmente correlacionados con la eficacia y la toxicidad de las pautas oncológicas son aclaramiento corporal total, el AUC, la concentración plasmática en estado estacionario (Cpss), y el tiempo durante el cual la concentración plasmática del fármaco se sitúa por encima de un determinado valor umbral. La mayoría de las correlaciones establecidas con estos parámetros hacen referencia a la toxicidad, ya que su medida es mucho más fácilmente cuantificable y objetivable que la medida de la eficacia clínica, que, además, generalmente suele ser mucho más diferida en el tiempo (tabla 3). En cualquier caso, las dependencias encontradas no son superiores al 70% de la respuesta evaluada; es decir, hay cerca de un 30% imputable a otros factores no esclarecidos (15). El AUC es el parámetro farmacocinético más frecuentemente correlacionado con la eficacia y/o toxicidad de los antineoplásicos. Sin embargo, en la bibliografía se detectan deficiencias en su cálculo que pueden condicionar la optimización de la pauta posológica seleccionada (1) (tabla 4).
Tabla 3. Parámetros farmacocinéticos correlacionados con la eficacia y toxicidad de antineoplásicos | ||||
| Fármaco | Correlación | Tipo de toxicidad | ||
| Eficacia | Toxicidad | |||
| Agentes alquilantes: | ||||
| -- Melfalan | -- | AUC | Renal. | |
| -- Busulfan | -- | AUC | Enfermedad venooclu siva. | |
| Complejos de platino: | ||||
| -- Cisplatino: total | Cl, AUC | Cl, AUC | Renal, digestiva y audi tiva. | |
| ultrafiltrado | AUC | Cl, AUC | Renal. | |
| -- Carboplatino | -- | AUC | Hematológica. | |
| -- Ipoplatino | -- | AUC | Hematológica. | |
| Antimetabolitos: | ||||
| -- Citarabina | Cl | -- | -- | |
| -- Metotrexato | AUC | Cl | Hematológica. | |
| -- Trimetrexate | -- | Cl, AUC | Hematológica. | |
| -- 5-fluorouracilo | AUC | AUC | Digestiva, hematoló gica y cardíaca. | |
| Antatriciclinas: | ||||
| -- Doxorrubicina | Cl, AUC | Css | Hematológica. | |
| -- Epirrubicina | AUC | AUC | Hematológica. | |
| -- Pirarrubicina | -- | AUC | Hematológica. | |
| -- Iododoxorrubicina | -- | AUC | Hematológica. | |
| -- Leurrubicina | -- | AUC | Hematológica. | |
| Epipodofilotoxinas: | ||||
| -- Etopósido | AUC | Css, AUC | Hematológica. | |
| -- Tenipósido | AUC | AUC | Hematológica. | |
| Alcaloides de la Vinca: | ||||
| -- Vinblastina | AUC | Css | Hematológica. | |
| -- Vincristina | AUC | AUC | Neurológica. | |
| Cl: aclaramiento plasmático. AUC: área bajo la curva. Css: concentración en el estado de equilibrio estacionario. Tomada de Desoise et al (12). | ||||
Tabla 4. Descripción de los métodos para el cálculo del área bajo la curva | |
Método | % frecuencia |
| -- Cálculo por trapezoidales | 70 |
| * Extrapolación al infinito | 67 |
| * Descripción del tiempo de perfusión | 64 |
| * Muestras a tiempo mayor de 3 t1/2 | 52 |
| -- Cálculo mediante ajuste exponencial del a curva | 13 |
| * Descripción del tiempo de perfusión | 33 |
| * Muestras a tiempo mayor de 3 t1/2 | 50 |
| -- Imposibilidad de evaluar el método de cálculo | 17 |
| Tomada de Siderov et al (1). | |
Además de los factores farmacocinéticos antes mencionados, el método de administración utilizado alcanza un papel relevante en el escenario de la optimización de dosis en oncología. Un ejemplo de esta situación es el 5FU, que actualmente continúa siendo el tratamiento estándar del cáncer colorrectal avanzado y, a pesar de su amplia utilización en los últimos quince años, todavía existe controversia en lo referente al método de administración. En efecto, al tratarse de un fármaco de fase S, con una semivida biológica muy corta, la administración en perfusión intravenosa continua permitiría una mayor exposición de las células tumorales al fármaco. Además, la administración en forma de bolus inhibe la síntesis de RNA de la célula tumoral sensible y la administración en perfusión continua provoca la inhibición de la timidilato sintetasa, mecanismo de acción de mayor citotoxicidad celular. Sin embargo, ha sido recientemente cuando se ha demostrado, mediante un meta-análisis, la superioridad de la perfusión continua frente a la perfusión intermitente en la supervivencia de los pacientes (16).
La monitorización de las concentraciones plasmáticas de los antineoplásicos como estrategia de individualización de su dosis es, desafortunadamente, mínima en la mayoría de los hospitales, a pesar de estar bien definida su indicación. En efecto, las premisas para la monitorización de forma rutinaria de las concentraciones plasmáticas de los agentes antineoplásicos han sido estructuradas por Grochow en 1998 (tabla 5) (2). Actualmente estas premisas se encuentran validadas para el 5FU, metotrexato a altas dosis (HDMTX), etopósido, tenipósido, citarabina, busulfán y carboplatino, ya que su aplicación en clínica ha presentado resultados prometedores (17). Un ejemplo claro de esta situación son los resultados de la monitorización de las concentraciones plasmáticas de metotrexato, tenipósido y citarabina para su individualización posológica en pacientes pediátricos diagnosticados de leucemia aguda linfoide (Fig. 3) (18). La monitorización garantiza un mayor porcentaje de ciclos con concentraciones plasmáticas dentro de los ámbitos terapéuticos definidos que cuando el tratamiento se aplica utilizando los criterios tradicionales y las dosis estándar.
Tabla 5. Premisas para la monitorización de las concentraciones plasmáticas de antineoplásicos |
| -- Imposibilidad de utilizar la respuesta clínica al tratamiento como herramienta básica para el ajuste individualizado de dosis debido a la dificultad en su medida. |
| -- Disponibilidad de técnicas analíticas con suficiente exactitud, precisión, especificidad y sensibilidad para la determinación de los agentes antineoplásicos y/o sus metabolitos. |
| -- Amplia variabilidad en los procesos farmacocinéticos de absorción, distribución y, fundamentalmente, eliminación por ser ésta la principal causa de la variabilidad en la exposición sistémica al antineoplásico. |
| -- Reducción en la variabilidad del efecto farmacológico al disminuir la variabilidad en la exposición sistemática al antineoplásico. |
| -- Mejor correlación entre concentración plasmática y efecto farmacológico que entre dosis y respuesta. |
| -- Validación de los principios farmacocinéticos y farmacodinámicos utilizados para la individualización posológica sobre la base del incremento de la efectividad del tratamiento, la reducción de la morbimortalidad asociada y la reducción del coste por proceso. |
| Adaptada de Growchow et al (2). |
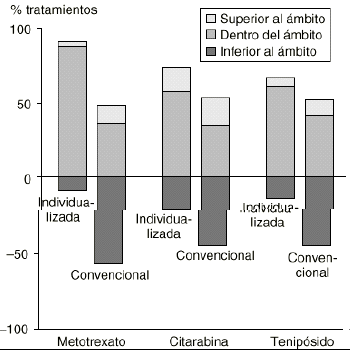
Figura 3.--Porcentajes de ciclos de tratamiento con concentraciones plasmáticas de metotrexato, tenipósido y citarabina mayores, iguales o menores al ámbito terapéutico definido en 91 pacientes con individualización de dosis y otros 91 pacientes con tratamiento convencional. Tomada de Evans et al (16).
El escaso desarrollo e implantación clínica de la monitorización de las concentraciones plasmáticas tienen su origen en causas multifactoriales. De éstas, la limitación de información en la descripción del método analítico en las publicaciones científicas se manifiesta como una de las más importantes (1) (tabla 6).
Tabla 6. Calidad de las publicaciones sobre las técnicas analíticas para la determinación de citostáticos | ||
Frecuencia (%) | ||
| -- Descripción de la preparación de la muestra y del material empleado | 77 | |
| -- Validación del método: | ||
| * Detalles de la curva de calibración | 10 | |
| * Exactitud | 13 | |
| * Precisión | 43 | |
| * Sensibilidad | 53 | |
| * Recuperación | 17 | |
| * Calidad de las muestras de control | 13 | |
| * Estabilidad del fármaco en plasma | 13 | |
| Tomada de Siderov et al (1). | ||
Los factores farmacodinámicos se han correlacionado, mayoritariamente, con la respuesta tóxica de los tratamientos, del mismo modo que las concentraciones plasmáticas de los antineoplásicos. En cualquier caso, la utilización en los modelos matemáticos de ajuste individualizado de dosis que considere parámetros farmacodinámicos, conjuntamente con la información farmacocinética, incrementa de forma notable la utilidad clínica de esta metodología. Así, la modelización del nadir de células blancas para la administración por vía oral de 50 mg/m2/día de etopósido en pacientes con cáncer no microcítico de pulmón en estadios IIIb o IV, evidencia la existencia de pacientes que se beneficiarían de un aumento de dosis, sin provocar aumento de la toxicidad, y la de pacientes en los que sería necesario una reducción de dosis para evitar la leucopenia grado IV (19) (tabla 7). El modelo desarrollado tiene la siguiente expresión:
WBCn = 0,35 · (1 WBCp · e1,12 · Cp)
Tabla 7. Concentraciones de etopósido asociadas con leucopenia grado IV tras el primer ciclo de quimioterapia en función del recuento de leucocitosal inicio del tratamiento | ||
| Percentil | Leucocitos prequimioterapia (*103/Ul) | Concentración plasmática en estado estacionario (ng/ml) |
| 0 | 3,4 | 0,87 |
| 25 | 7,9 | 1,33 |
| 50 | 9,4 | 1,42 |
| 75 | 13,4 | 1,62 |
| 100 | 17,2 | 1,72 |
Tomada de Miller et al (19). | ||
donde WBCn es el nadir de leucocitos, WBCp es el recuento de leucocitos pretratamiento y Cp es la concentración plasmática en situación de estado estacionario. Del mismo modo, los modelos que a priori incorporan la reducción deseada del número de plaquetas tras la administración del carboplatino se han mostrado útiles en la racionalización de las pautas posológicas de este fármaco (tabla 2).
En resumen, para el ajuste individualizado de dosis la mayor o menor consideración de los parámetros antropométricos, fisiológicos, farmacocinéticos y farmacodinámicos, es dependiente de su aportación contrastada, mediante ensayos clínicos, a la reducción de la variabilidad en la respuesta de los fármacos antineoplásicos.
MÉTODOS DE AJUSTE INDIVIDUALIZADO DE DOSIS Y SU VALIDACIÓN
El establecimiento de la pauta posológica estándar de un fármaco, clásicamente, se ha basado en realización de estudios dosis-respuesta y, más recientemente, en estudios farmacocinéticos-farmacodinámicos realizados durante las fases iniciales del desarrollo de los nuevos fármacos. Sin embargo, en las fases I y II el fármaco se administra a una población de pacientes cuyas características fisiopatológicas frecuentemente son distintas de la población a la cual va a ser administrado cuando se comercialice. Este hecho parece confirmarse también para los antineoplásicos, puesto que de los cerca de 9.000 ensayos clínicos publicados en inglés y registrados en Medline en los últimos diez años (1988-1997) sobre fármacos antineoplásicos, tan sólo 350 (3-4%) hacían referencia a su comportamiento farmacocinético y menos de 50 (<1%) incluyen estudios farmacocinéticos y farmacodinámicos. Esta situación sorprende por cuanto que las ventajas que presentan este tipo de estudios están suficientemente contrastadas y difundidas (20) (tabla 8).
Tabla 8. Ventajas de los estudios farmacocinéticos-farmacodinámicos en oncología |
| -- Determinar del perfil farmacocinético del fármaco en poblaciones de pacientes representativos de la patología a tratar (población diana). |
| -- Cuantificar la variabilidad interpaciente y su relación con los factores fisiopatológicos (modelo farmacocinético poblacional). |
| -- Calcular los parámetros farmacocinéticos de los pacientes de forma individual utilizando el modelo poblacional y/o estimación bayesiana (ajuste individualizado de dosis). |
| -- Investigar el valor pronóstico de los parámetros farmacocinéticos individuales en relación a los resultados clínicos (parámetros farmacodinámicos) predefinidos en términos de eficacia y seguridad (modelo farmacocinético-farmacodinámico). |
| Adaptada de Bruno et al (20). |
En este contexto podría argüirse la escasa implantación de la individualización posológica en oncología, basada en estudios farmacocinéticos y farmacodinámicos es, posiblemente, consecuencia no sólo de las distintas formas de abordar el problema, sino, además, de la dificultad inherente a la misma, derivada de la existencia de diferentes métodos de ajuste individualizado de dosis para un mismo protocolo quimioterápico y/o enfermedad cancerígena, y de la distinta magnitud de la variabilidad intraindividual e interindividual de cada citostático.
En el caso de citostáticos cuya variabilidad intraindividual sea menor que la variabilidad interindividual es posible estudiar el comportamiento cinético del citostático en el primer ciclo e individualizar la dosis para los ciclos sucesivos. El parámetro farmacocinético de mayor utilidad clínica demostrada para el ajuste individualizado de dosis es el AUC y/o el aclaramiento plasmático. No obstante, su correcta cuantificación por métodos farmacocinéticos no compartimentales requiere un elevado número de muestras plasmáticas que no es posible obtener en clínica asistencial. La resolución de este problema se ha abordado mediante la aplicación de los modelos de estimación del AUC a partir de un número de muestras limitadas (LSM, acrónimo de limited-sampling models, en inglés) y cuando el comportamiento farmacocinético poblacional se encuentra caracterizado, mediante la aplicación de la metodología bayesiana.
Los modelos de estimación del AUC han sido desarrollados y validados para varios citostáticos (tabla 9). El desarrollo del modelo se realiza mediante un análisis multivariante a través de regresión lineal múltiple, no condicional, en pasos sucesivos. En este análisis la variable dependiente es el AUC, calculada por métodos no compartimentales o compartimentales, y las variables independientes son las concentraciones de fármaco extraídas a distintos tiempos de muestreo y las dosis administradas cuando el AUC no se encuentre normalizado por la dosis. Estos modelos requieren validación previa a su implementación en clínica. Este proceso, iterativo y no secuencial, se lleva a cabo determinando la exactitud (el error relativo medio de predicción) y precisión (raíz cuadrada del error cuadrático medio de predicción) de las estimas calculadas con los mismos mediante la teoría de los errores de predicción de Sheiner y Beal (21).
Tabla 9. Fármacos antineoplásicos con modelo de estimación del AUC. | ||
| Fármaco | Referencia | |
| Agentes alquilantes: | ||
| -- Ciclofosfamida | Egorin, 1989 (51) | |
| -- Tiotepa | Ackland, 1988 (52) | |
| Complejos de platino: | ||
| -- Cisplatino | Desoize, 1992 (54) | |
| -- Carboplatino | Sorensen, 1993 (55) | |
| Antimetabolitos: | ||
| -- Metotrexato | Iliadis, 1985 (58) | |
| -- 5-fluorouracilo | Santini, 1989 (59) | |
| -- Cladribina | Liliemark, 1996 (60) | |
| Antraciclinas: | ||
| -- Doxorrubicina | Ratain, 1989 (24) | |
| -- Epirrubicina | Eksborg, 1990 (53) | |
| Derivados de camptotecina | ||
| -- Topotecan | Van Warmerdam, 1996 (56) | |
| -- Irinotecan | Mick, 1996 (57) | |
| Alcaloides de la Vinca: | ||
| -- Vinblastina | Ratain, 1987 (61) | |
| Epipodofilotoxinas: | ||
| -- Etopósido | Mick, 1991 (62) | |
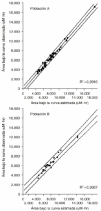
En nuestro hospital se han desarrollado modelos de estimación del AUC para la administración de HDMTX en pacientes diagnosticados de osteosarcoma y altas dosis de ciclofosfamida en pacientes diagnosticados de cáncer de mama y subsidiarios de quimioterapia mieloablativa y rescate con células progenitoras hematopoyéticas. En el caso del metrotexato, el análisis multivariante evidencia cómo las concentraciones plasmáticas extraídas durante la fase de distribución rápida (es decir, a los diez minutos, tres y doce horas postperfusión) poseen una mayor capacidad explicativa de la variabilidad asociada al AUC que el resto de concentraciones plasmáticas (tabla 10). La validación del modelo LSM para la administración del metrotexato a altas dosis se muestra en la tabla 11 (22). Los resultados del coeficiente de correlación en la predicción del AUC a partir de las concentraciones plasmáticas, tanto en la población de validación (B) como en la de referencia (A), ponen de manifiesto la excelente exactitud y precisión del modelo desarrollado. En efecto, el intervalo de confianza del error relativo medio carece de sesgo sistemático (p > 0,05) al incluir el valor 0 en su ámbito de valores, y la raíz cuadrada del error cuadrático medio es inferior al 15%, valor umbral establecido como precisión mínima exigible para los modelos de estimación del AUC. Estos resultados se confirman al representar el diagrama de dispersión de los valores de AUC obtenidos con el modelo de estimación a partir de un número reducido de muestras plasmáticas y los valores de AUC determinados con los parámetros farmacocinéticos obtenidos de cada ciclo, correspondientes a la población A y a la población B (Fig. 4).
Tabla 10. Cálculo del área bajo la curva (AUC) de ciclofosfamida y metotrexato mediante modelos de estimación del AUC | |||||
Fármaco | Dosis (g/m2) | Perf. IV (h) | AUC | r2 | Validación |
| Ciclofosfamida* | 6 | 96 | 755,7 + 24,7 · C90 h + 27,9 · C72 h + 23,8 · C48 h | 0,87 | No |
| Metotrexato (22) | 12 | 6 | 489,4 + 1,9 C10'' + 10,4 C3 h + 13,3 Cp12 h | 0,98 | Sí |
Tabla 11. Validación del modelo de estimación del AUC del metotrexato administrado a altas dosis | ||||
| Población | Ptes/ciclos | r2 | ER (%) | RECM (%) |
| A | 5/51 | 0,981 (0,968-0,989) | 0,26 (1,35, 1,88) | 4,46 3,46, 5,47) |
| B | 6/23 | 0,967 (0,922-0,986) | 1,50 (4,50, 0,48) | 6,93 (4,47, 8,72) |
*Datos no publicados. | ||||
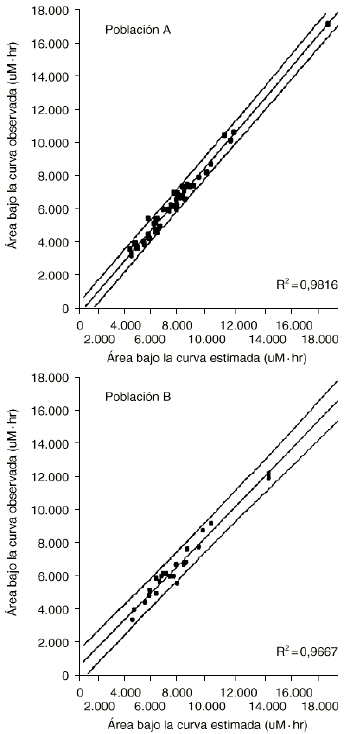
Figura 4.--Área bajo la curva calculada a partir de los parámetros farmacocinéticos (AUC observada) y área bajo la curva calculada a partir del modelo de estimación del AUC a partir de un número de muestras limitada (AUC estimada) en las poblaciones A y B (en ambos casos se han dibujado las líneas de regresión y sus IC95% asociados a las predicciones individuales). Tomada de Peris et al (22).
En el caso de la ciclofosfamida las concentraciones plasmáticas extraídas a las cuarenta y ocho, setenta y dos y noventa horas después del inicio de la perfusión son óptimas para la estimación del AUC. Esto es justo donde el cambio de las concentraciones plasmáticas es debido, mayoritariamente, a la autoinducción metabólica que ocurre tras las primeras veinticuatro horas de la administración en perfusión intravenosa continua de este fármaco (23).
El creciente desarrollo de los modelos de estimación del AUC para diferentes citostáticos se debe a su sencillez, pero exige que la toma de muestras en sangre se realice exactamente a los tiempos establecidos y sólo es aplicable para aquellas dosis en las que se ha desarrollado y validado el mismo. Esta circunstancia restringe la utilización de estos modelos cuando no se realiza su validación (24). Un ejemplo de este hecho se evidencia en la tabla 12, donde los cambios en la pauta posológica del etopósido condicionan la selección de los tiempos para la estimación del AUC. Estas condiciones tan restrictivas no se presentan con la caracterización de los parámetros farmacocinéticos individuales mediante la metodología bayesiana al tratarse de modelos estocásticos (probabilísticos) y no de modelos determinísticos como son los modelos de estimación del AUC.
Tabla 12. Cálculo del área bajo la curva (UAC) de etopósido mediante modelos de estimación del UAC basado en un número reducido de concentraciones plasmáticas | |||||
| Dosis (mg/m2) | Perf. IV (h) | AUC (mg/ml · min.) | r | Validado | Referencias |
| 100 | 2 | 3,3 · C1,5 h + 12,7 · C10 h + 32,0 · C24 h 3,8 | 0,95 | Sí | Joel, 1990 (63) |
| 150 | 1 | 0,004 · C2 h + 0,007 · C4 h + 0,02 | NI | No* | Miller, 1992 (64) |
| 75-150 | 1,5 | 0,001 · D + 766 C4 h | 0,92 | Sí | Strömgren, 1993 (65) |
| 80-150 | NI | 0,343 · C4 h + 0,650 · C8 h + 1,252 | 0,96 | Sí | Holz, 1995 (66) |
| 100** | -- | 0,631 · C4 h + 0,336 · C6 h + 0,376 | 0,99 | Sí | Millward, 1995 (67) |
*Modelos de toma de muestra limitado con validación no satisfactoria. **Dosis expresada como mg/día y administrada por vía oral. NI: no indicado. | |||||
Sin embargo, la utilidad de la caracterización del comportamiento cinético de un fármaco mediante esta metodología puede carecer de interés clínico cuando la variabilidad en los parámetros farmacocinéticos de un individuo, entre los distintos ciclos de quimioterapia que recibe, iguala o supera la variabilidad interindividual. En consecuencia, cuando la variabilidad se manifiesta en este sentido obligatoriamente hay que hacer seguimiento farmacocinético individual en cada ciclo. Si el paciente recibe más de un citostático se debe seleccionar el más crítico o el de mayor variabilidad de la pauta o protocolo, y se valida si su monitorización plasmática proporciona información suficiente para la individualización posológica; en caso contrario, se han de incluir en el programa de monitorización todos los antineoplásicos del protocolo (25).
Una posible estrategia para abordar este problema consiste en la administración de una dosis previa seguida de la monitorización intensiva de las concentraciones plasmáticas del fármaco con el objetivo de caracterizar los parámetros farmacocinéticos del paciente de forma individual y predecir la dosis a administrar posteriormente. Este método ha sido ampliamente utilizado en la individualización del metotrexato a altas dosis (26).
Sin embargo, el método bayesiano para la individualización posológica posee mayor eficiencia que la administración de una dosis previa, seguida de la monitorización intensiva de las concentraciones plasmáticas del fármaco. La aproximación bayesiana, basándose en la expectativa a priori y en el conocimiento de la variabilidad con que se da dicha expectativa, permite una visión más intuitiva acerca del paciente. Este método de optimización de dosis es más eficaz, pero de mayor complejidad que los modelos de estimación del AUC al requerir la correcta caracterización de los parámetros farmacocinéticos poblacionales tanto de tendencia central como de variabilidad (19), y la optimización del diseño experimental, puesto que la estimación eficiente de los parámetros individuales va a depender de los tiempos de muestreo, que a su vez depende de los parámetros farmacocinéticos poblacionales de tendencia central y de su variabilidad (27).
Además esta aproximación puede ser utilizada tanto cuando la variabilidad en los parámetros farmacocinéticos de un individuo, entre los distintos ciclos de quimioterapia que recibe, iguala o supera la variabilidad interindividual como cuando la variabilidad intraindividual sea menor que la variabilidad interindividual. El metotrexato es un fármaco de estrecho ámbito terapéutico y amplia variabilidad intraindividual (superior a la variabilidad interindividual) que exige la monitorización de sus concentraciones plasmáticas y el establecimiento de un conjunto de acciones dirigidas a prevenir su grave toxicidad potencial sin interferir en su actividad antitumoral en cada ciclo que recibe el paciente. A pesar de ello no menos del 20% de los ciclos, con dosis altas de metotrexato, se manifiestan de alto riesgo. En el tratamiento neoadyuvante del osteosarcoma con metotrexato los resultados clínicos obtenidos prueban cómo la supervivencia libre de enfermedad se incrementa a medida que los pacientes reciben mayor intensidad de dosis de metotrexato (28). Esta información es consistente con la necesidad de monitorizar las concentraciones plasmáticas a fin de individualizar las dosis de metotrexato a administrar al paciente en cada ciclo para conseguir concentraciones plasmáticas pico del orden de 1.000 µM (Fig. 5). Este criterio posológico, soportado por la metodología bayesiana, ha supuesto el abandono del peso y/o la superficie corporal del paciente como variables para determinar la dosis individualizada de metotrexato en nuestro hospital. En la práctica la aplicación de este criterio representa trasladar la mayor parte de la variabilidad intraindividual en la concentración pico a la dosis administrada en los pacientes (Fig. 6).
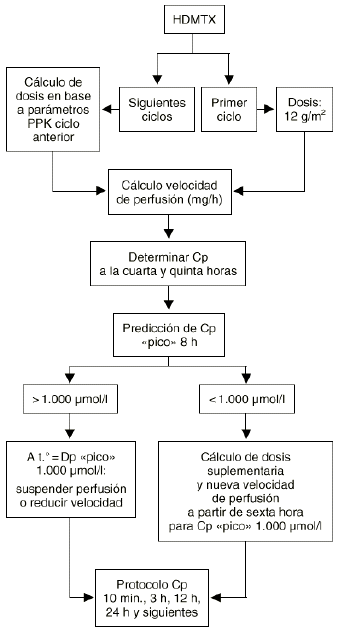
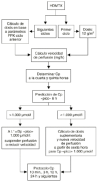
Figura 5.--Diagrama utilizado en el Hospital Universitario Dr. Peset para la individualización de altas dosis i.v. de MTX en pacientes diagnosticados de osteosarcoma y obtención de una concentracion plasmática pico de 1.000 µmol/l. Tomada de Jiménez et al (68).
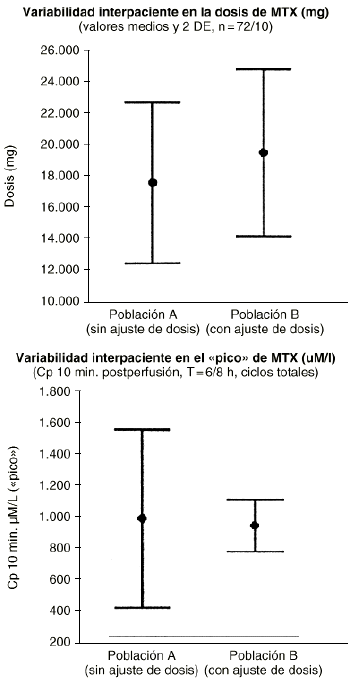
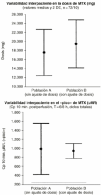
Figura 6.--Influencia del método de ajuste posológico (gm/m2 versus concentración diana >= 1.000 µmoles/litro) de metotrexato a altas dosis sobre la variabilidad en la dosis (g/m2) y en la concentración (µmoles/ml) interciclo.
Por otra parte, el 5-fluorouracilo es un fármaco cuya variabilidad intraindividual es menor que la variabilidad interindividual. El modelo bayesiano desarrollado para predecir el comportamiento cinético individual del 5-Fu, utilizando las concentraciones plasmáticas obtenidas durante el primer ciclo de tratamiento (10), evidencia que únicamente el conocimiento de la concentración plasmática de los diez minutos postperfusión (Cp1) mejora la exactitud (expresada como error absoluto) del modelo, de forma estadísticamente significativa, con respecto a las predicciones realizadas a priori y a posteriori (10) (Fig. 7). Por tanto, disponer de las muestras a los treinta y sesenta minutos postperfusión (Cp2 y Cp3), conjuntamente a la concentración plasmática a los diez minutos (Cp1) de finalizada la perfusión, no ofrece ventajas para la individualización posológica de ciclos sucesivos por cuanto que no mejora la información proporcionada por el modelo a priori desarrollado tanto con el programa PKS como con NONMEM. Esta metodología ha sido validada para otros citostáticos como son el etopósido (29), el carboplatino (50) y el cisplatino (54), entre otros.
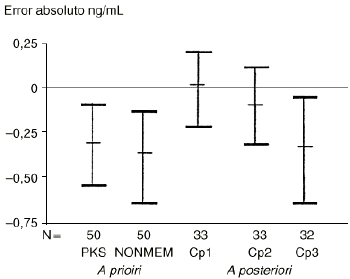
Figura 7.--Validación de los tiempos de toma de muestra para el 5FU en dosis única. Tomada de Climente (10).
La individualización posológica de los tratamientos antineoplásicos puede ser de mayor importancia cuando se administra quimioterapia de intensificación. Desde que Hriniuck (30) en 1988 introdujo el concepto de intensidad de dosis, presentando los resultados de una serie de estudios retrospectivos que avalaban la importancia de este concepto en el tratamiento del cáncer de mama, numerosos investigadores han demostrado la importancia de este procedimiento en la respuesta de los pacientes con cáncer de mama, linfomas no Hodgkin, cáncer microcítico del pulmón, osteosarcoma y otros más (31-33). Ahora bien, como la conocida frase more is better (34) estaba sustentada sobre estudios retrospectivos o estudios pilotos no aleatorizados, si no se quiere comprometer el máximo beneficio en el paciente, la expresión debería referirse como «más es mejor, pero no mucho más es mucho mejor» (35). En consecuencia, la individualización posológica de los pacientes basada en la caracterización del perfil cinético individual puede llegar a ser tan importante como la aplicación del tratamiento. Esta importancia estriba en que la variabilidad de los parámetros AUC y Cl, que determinan, respectivamente, la cantidad y la duración de la exposición sistémica del organismo al citostático, está influenciada por covariables de difícil cuantificación y, a veces, de difícil identificación. En efecto, el metotrexato, etopósido, tenipósido, doxorrubicina, ciclofosfamida, bleomicina, entre otros citostáticos, presentan un aclaramiento en pacientes oncológicos, adultos y niños, con diferencias afectadas por coeficientes entre 2 y 10 (36).
En lo referente a la quimioterapia mieloablativa, previa reinfusión de células periféricas, y pese a la avalancha de datos publicados en la última década, no existen suficientes ensayos clínicos aleatorizados en cáncer de mama, en general, que las avalen; además, los resultados no son tan nítidos como para no precisar este refrendo (37). De hecho, la quimioterapia de intensificación con carácter adyuvante en pacientes con cáncer de mama de alto riesgo no es hoy día una indicación aprobada por la Dirección General de Farmacia y Productos Sanitarios, y aunque sea posiblemente útil no está suficientemente documentada.
Finalmente, un ejemplo paradigmático de la individualización posológica en la quimioterapia antineoplásica lo podría representar el tratamiento integral de la leucemia aguda linfoblástica (LAL) en la población pediátrica, enfermedad curable en el 70% de los casos (38), que incluye metotrexato, tenipósido y citarabina. Evans et al (18) en un estudio prospectivo con 188 niños asignados de forma aleatoria al tratamiento convencional (basado en la superficie corporal) o individualizado (basado en un CI y un AUC predefinidos) encuentran a los cinco años que el 72 (6)% del grupo de tratamiento individualizado frente al 66 (6)% para el grupo de pacientes en tratamiento convencional continúan en remisión completa (p < 0,05).
En nuestra opinión y de acuerdo con el estado actual de la individualización posológica en oncología, basada en la utilización de modelos farmacocinéticos farmacodinámicos, plantear un programa de estas características para todos los antineoplásicos y en cada ciclo que recibe el paciente es una situación ideal y que en cualquier caso únicamente se debe realizar cuando se haya demostrado el beneficio previo en los pacientes. Ahora bien, mientras se alcanza esta situación la búsqueda de alternativas es un reto de todos los profesionales sanitarios con implicación directa en el cuidado de los pacientes oncológicos y en el uso racional de los medicamentos citostáticos. En definitiva, existe la necesidad de potenciar la investigación farmacocinética-farmacodinámica en oncología para reducir la morbimortalidad asociada al tratamiento farmacológico del cáncer.
BIBLIOGRAFÍA
1. Siderov J, Brien JE, Morgan DJ, Zalcberg J, Coloso W. Quality of pharmacokinetic research in oncology. Br J Cancer 1995;72:792-4.
2. Grochow LB. Individualized dosing of anti-cancer drugs and the role of therapeutic monitoring. En: Grochow L, Ames M, eds. A clinician''s guide to chemotherapy pharmacokinetics and pharmacodynamics, 1.a ed. USA: Williams & Wrzosek; 1998. p. 3-53.
3. Gurney H. Dose calculation of anticancer drugs: a review of the current practice and introduction of an alternative. J Clin Oncol 1996;14:2590-611.
4. Holford NHG. Input from the deep south compartment. A personal viewpoint. Clin Pharmacokinet 1995;29:139-41.
5. Holford NHG, Sheiner LB. Kinetics of pharmacologic response. J Pharmal Ther 1982;16:143-66.
6. Ling V, Charles F. Kettering price: P-glycoprotein and resistance to anticancer drugs. Cancer 1922;69:2603-9.
7. Wacher WJ, Wu CY, Benet LZ. Overlapping substrate specificities and tissue distribution of cytochrome P450 3a and glycoprotein P: implications for drug delivery and activity in cancer chemotherapy. Mol Carcinog 1995;13:129-34.
8. Bacci G, Ferrari S, Delepine N, et al. Predictive factors of histiologic response to primary chemotherapy in osteosarcoma of the estremity: study of 272 patients preoperatively treated with high-dose methothrexate, doxorrubicin, and cisplatin. J Clin Oncol 1998;16:658-63.
9. Dufull S, Robinson BA. Clinical pharmacokinetics and dose optimisation of carboplatin. Clin Pharmacokinet 1997;33:161-82.
10. Climent M. Farmacocinética poblacional de 5-fluoracilo en pacientes con cáncer colorrectal [tesis doctoral]. Valencia: Universidad de Valencia. Facultad de Farmacia; 1997.
11. Gurney HP, Ackland S, Gebski V, Farrell G. Factors affecting epirrubicin pharmacokinetics and toxicity: evidence against using body-surface area for dose calculation. J Clin Oncol 1998;16:2299-304.
12. Calvert AH, Newel DR, Gumbrell LA, et al. Carboplatin dosage: prospective evaluation of a simple formula based on renal function. J Clin Oncol 1989;7:1748-56.
13. Launay-Iliadis MC, Bruno R, Cosson V, Vergniol JC, Oulid-Aissa D, Marty M, Clavel M, Aapro M, Le Bail N, Iliadis A. Population pharmacokinetics of docexatel during phase I studies using nonlinear mixed-effect modeling and nonparametric maximum-likelihood estimation. Cancer Chemother Pharmacol 1995;37:47-54.
14. Bruno R, Vivier N, Vergiol JC. A population pharmacokinetic model for doxatel (taxotere): Model building and validation. J Pharmacokinet Biopharm 1996;24:153-72.
15. Desoize B, Robert J. Individual dose adaptation of anticancer drugs. Eur J Cancer 1994;30:844-51.
16. The meta-analysis group in cancer. Efficacy of intravenous continuous infusion of fluorouracil compared with bolus administration in advanced colorectal cancer. J Clin Oncol 1998;16:301-8.
17. Plunkett W, Gandhi V. Cellular pharmacodynamics of anticancer drugs. Semin Oncol 1993;20:50.
18. Evans W, Relling V, Rodman J, Crom W, Bonett J, Pui C. Convencional compared with individualized chemotherapy for childhood acute lymphoblastic leukemia. N Engl J Med 1998;338:499-505.
19. Miller A, Tolley E. Predictive performance of a pharmacodynamic model for oral etoposide. Cancer Res 1994;54:2080-3.
20. Bruno R, Hille D, Riva A, et al. Population pharmacokinetics/pharmacodynamics of doxatel in phase II studies in pacients with Cancer. J Clin Oncol 1998; 16:187-96.
21. Sheiner LB, Beal SL. Some suggestions for measuring predictive performance. J Pharmacokinet Biopharm 1981;9:503-12.
22. Peris JF, Pérez JJ, Alós M, Jiménez NV. Optimización de la toma de muestra para estimar el área bajo la curva de metotrexato en pacientes con osteosarcoma. XLI Congreso de la Sociedad Española de Farmacia Hospitalaria. Sevilla; 1996. p. 305-6.
23. Chen TL, Passos-Coelho M, Noe DA, et al. Nonlinear pharmacokinetics of cyclophosphamide in patients with metastasic breast cancer receiving high dose chemotherapy followed by autologous bone marrow trasplantation. Can Res 1995;55:810-6.
24. Warmerdam L, Bokkel W, Maes R, Beijnen J. Limited-sampling models for anticancer agents. J Cancer Res Clin Oncol 1994;120:427-33.
25. Sandström M, Freijs A, Larsson R, Nygren P, Fjällskog MI, Bergh J, Karlsson. Lack of relationship between systemic exposure for the component drugs of the fluorouracil, epirubicin, and 4-hydroxycyclofosfamide regimen in breast cancer pacients. J Clin Oncol 1996;14:1581-8.
26. Bruno R, Iliadis A, Favre R, Lena N, Imbert AM, Cano JP. Dosage predictions in high-dose metrotrexate infusions. Part 2: Bayesian estimation of methotrexate clearance. Cancer Drug Delivery 1985;2:277-83.
27. Pérez-Ruixo JJ, Medina C, Casabó VG, Jiménez NV. Influencia del diseño experimental en la estimación de parámetros farmacocinéticos poblacionales de ciclofosfamida en pacientes con cáncer de mama. Farm Hosp 1998;22:9-15.
28. Delepine N, Delepine G, Bacci G, Rosen G, Debois J. Influence of metrotrexate dose intensity on outcome of patients with high grade osteogenic osteosarcoma. Cancer 1996;78:2127-34.
29. Ratain MJ, Shilsky RL, Choi KE. Adaptive control of etoposide dosing: impact of inter pacient pharmacodynamic variability. Clin Pharmacol Ther 1989;45: 226-33.
30. Hriniuk WM. The importance of dose intensity in the outcome of chemotherapy. Important Avd Oncol 1988;3:121-41.
31. Carmo-Pereira J, Costa FO, Henriques E. A compararison of two doses of adriamycin in the primary chemotherapy of disseminated breast carcinoma. Br J Cancer 1987;56:471-3.
32. Gurney H, Dodwell D, Thather N. Escalating drug delivery in cancer chemotherapy: a review of concepts and practice. Part 1. Ann Oncol 1993;4:23-34.
33. Gurney H, Dodwell D, Thather N. Escalating drug delivery in cancer chemotherapy: a review of concepts and practice. Part 2. Ann Oncol 1993;4:103-15.
34. Hriniuk WM. Morer is Better. J Clin Oncol 1988;6: 1365-7.
35. Wood WC, Budman DR, Korzun AH, Cooper B, Younger J, Hart R. Dose and dose intensity of adjuvant chemotherapy for stage II, node positive breast cancer. N Engl J Med 1994;330:1253-9.
36. Evans WE, Relling MV. Clinical pharmacokinetics-pharmacodynamics of anticancer drugs. Clinical Pharmacokinetics 1989;16:327-36.
37. Gore M, Mainwaring P, A''Hern R, et al. Randomized trial of dose-intensity with single agents carboplatin in pacients with epithelial ovarian cancer. J Clin Oncol 1998;16:2426-34.
38. Evans WE, Crom WR, Abromowith M. Clinical pharmacokinetics and pharmacodynamics of anticancer drugs in children. Semin Oncol 1993;20:18-29.
39. Madden T, Sunderland M, Santana VM. The pharmacokinetics of high-dose carboplatin in pedriatric pacients with cancer. Clin Pharmacol Ther 1992;51:701-7.
40. Newell DR, Pearson AD, Balmanno K. Carboplatin pharmacokinetics in children: the development of a pedriatic dosing formula. The United Kingdom Children''s Cancer Study Group. J Clin Oncol 1993;11: 2314-23.
41. Cosolo WC, Morgan DJ, Seeman E. Lean body mass, body surface area and epirubicin kinetics. Anticancer Drugs 1994;5:293-7.
42. Lowis SP, Pearson AD, Newell DR. Etoposide pharmacokinetics in children: the development and prospective validation of a dosing equation. Cancer Res 1993;53:4881-9.
43. Ratain MJ, Mick R, Shilsky RL. Pharmacologically based dosing of etoposide: a means of safely increasing dose intensity. J Clin Oncol 1991;9:1480-6.
44. Body AV, Yule SM, Wyllie R. Pharmacodynamics and metabolism of ifosfamide administered as a continuous infusion in children. Cancer Res 1993;53: 3758-64.
45. Sonnichsen DS, Hurwitz CA, Pratt CB. Saturable pharmacokinetics and paclitaxel pharmacodynamics in children with solid tumors. J Clin Oncol 1994;12:532-8.
46. Grochow LB, Baraldi C, Noe D. Is dose normalization to weight or body surface area useful in adults? J Natl Cancer Inst 1990;82:323-5.
47. Egorin MJ, Van Echo DA, Tipping SJ et al. Pharmacokinetics and dosage reduction of cis-diammine(11-cyclobutanedi-carboxylato)platinum in patients with impared rebal function. Cancer Res 1984;44:5432-8.
48. Calvert AH, Newell DR, Gumbrell LA, et al. Carboplatin dosage: prospective evaluation of simple formula based on renal function. J Clin Oncol 1989;7:1748-56.
49. Chatelut E, Canal P, Brumner V, et al. Prediction of carboplatin clearance from standard morphological and biological patient characteristics. J Nat Cancer Inst 1995;87:573-80.
50. Dufull SB, Begg EJ, Robinson BA, et al. A sequential Bayesian algorithm for dose individualization of carboplatin. Cancer Chemother Pharmacol 1997;39:317-26.
51. Egorin MJ, Forrest A, Belani CP, Ratain MJ, Abrams JS, Echo DA. A limited sampling strategy for cyclophosphamide pharmacokinetics. Cancer Res 1989; 49:3129-33.
52. Ackland SP, Choi RE, Ratain MJ, Ergorin MJ, Williams SF, Bitran JD. Human plasma pharmacokinetics of thiotepa following high-dose administration of thiotepa and cyclophosphamide. J Clin Oncol 1988; 6:1192-6.
53. Eksborg S. Anthracycline pharmacokinetics. Limited sampling model for plasma level monitoring with special reference to epirubicin (Farmorubicin). Acta Oncol 1990;29:339-42.
54. Desoize B, Dufour R, Collery P, Unien S. Bayesian estimation of cisplatin pharmacokinetics during five-day continuous infusions. Metal Ions in Biology and Medicine 1992;2:182-3.
55. Sorensen BT, Strömgren A, Jakobsen P, Jakobsen U. A limited sampling method for estimation of carboplatin area under the curve. Cancer Chemother Pharmacol 1993;31:324-7.
56. Van Warmerdam LJC, Creemers GJ, Rodenhuis S, et al. Pharmacokinetics and pharmacodynamics of topotecan given on a daily-times-five schedule in phase II clinical trials using a limited-sampling procedure. Cancer Chemother Pharmacol 1996;38:254-60.
57. Mick R, Gupta E, Vokes EE, Ratain MJ. Limited-sampling models for irinotecan pharmacokinetics-pharmacodynamics: prediction of biliary index and intestinal toxicity. J Clin Oncol 1996;14:2012-9.
58. Idiadis A, Bachir-Raho M, Bruno R, Favre R. Bayesian estimation and prediction of clearance in high dose methotrexate infusions. J Pharmacokin Biopharm 1985;13:101-15.
59. Sataini J, Milano G, Thyss A. 5-Fu therapeutic monitoring with dose adjustment leads to improved therapeutic index in headf and neck cancer. Br J Cancer 1989;59:287-90.
60. Liliemark J, Albertioni F, Juliusson G, Eksborg S. A limited sampling strategy for estimation of the cladribine plasma area under the concentration versus time curve after intermittent i.v. infusion, s.c. injection, and oral administration. Cancer Chemother Pharmacol 1996;35:536-40.
61. Ratain MJ, Vogelzang NJ. A limited sampling for vinblastine pharmacokinetics. Cancer Treat Rep 1987; 71:935-9.
62. Mick AA, Ratain MJ. Modeling interpatient pharmacodynamic variability of etoposido. J Natl Cancer Inst 1991;83:1560-4.
63. Joel SP, Heap L, Robbins S, Clark PI, Slevin ML. A limited sampling strategy for the calculation of etoposido pharmacokinetic. Proc Am Assoc Clin Oncol 1990;9:67.
64. Miller AA, Tolley EA, Niell HB, Stewart CF, Griffin JP. Pharmacokinetics of three daily infusions of etoposide in pacients with extensive-stage small-cell lung cancer. Cancer Chemother Pharmacol 1992;31: 161-6.
65. Strömgren A, Sorensen BT, Jakobsen P, Jakobsen A. A limited sampling method for estimation of the etoposide area under the curve. Cancer Chemother Pharmacol 1993;32:226-30.
66. Holz JB, Kopper H, Schmidt L, Fritsch HW, Pfluger KH, Juglas H. Limited sampling models for reliable estimation etoposide area under the curve. Eur J Cancer 1995;31:1794-8.
67. Millward MJ, Newell DR, Yuen K, Matthews JP, Balmanno K, Charlton CJ, Gumbrell L, Lind MJ, Chapman F, Protor M. Pharmacokinetics and pharmacodynamics of prolonged oral etoposide in woman with metastatic breast cancer. Cancer Chemother Pharmacol 1995;37:161-7.
68. Jiménez NV, Casabó VG, Sancho V. Manual de procedimientos para farmacocinética clínica, 1.a ed. Valencia: AFAHPE, 1997. Capítulo IV.
Este artículo se presentó en forma de conferencia en el XXIII Congreso Internacional de la Sociedad Farmacéutica del Mediterráneo Latino celebrado en Toledo del 30 de septiembre al 2 de octubre de 1998.

