







Desarrollar y validar internamente un modelo farmacocinético poblacional para gemcitabina y su metabolito 2’,2’-difluorodeoxiuridina (dFdU) y evaluar su capacidad predictiva en la personalización de la dosificación de gemcitabina en pacientes oncológicos.
MétodosLas concentraciones plasmáticas de gemcitabina y dFdU se determinaron en 18 pacientes oncológicos. Se determinaron los parámetros farmacocinéticos correspondientes a un modelo farmacocinético bicompartimental mediante el programa NONMEN VI. La capacidad para identificar los parámetros se evaluó mediante bootstrap paramétrico. La validación interna del modelo se realizó mediante bootstrap no paramétrico, visual y numerical predictive check. La capacidad predictiva del modelo final se evaluó en términos de precisión y exactitud durante el primer (a priori) y segundo (a posteriori) ciclo de quimioterapia.
ResultadosEl valor medio y la variabilidad interpaciente del aclaramiento de gemcitabina y de dFdU es de 2,70 l/min (31,0%) y 0,0515 l/min (35,8%), respectivamente. El volumen de distribución de gemcitabina y dFdU estimado en estado equilibrio estacionario es de 30 y 238 l, respectivamente. La validación interna confirma que el modelo farmacocinético poblacional resulta adecuado para describir la evolución temporal de las concentraciones plasmáticas de gemcitabina y dFdU, así como su variabilidad en la población de estudio. La exactitud y la precisión a posteriori de las concentraciones plasmáticas de gemcitabina mejoran un 67% y un 46%, respectivamente, respecto a la predicción a priori.
ConclusiónEl modelo farmacocinético poblacional desarrollado caracteriza adecuadamente la evolución temporal de las concentraciones plasmáticas de gemcitabina y dFdU en la población de estudio y puede utilizarse de forma exacta y precisa para optimizar las pautas posológicas de gemcitabina en pacientes oncológicos.
To develop and internally validate a population pharmacokinetic model for gemcitabine and its metabolite 2’,2’-difluorodeoxyuridine (dFdU); and to evaluate its predictive perfomance for personalizing the dosage used in cancer patients.
MethodsGemcitabine and dFdU plasma concentrations were determined in 18 cancer patients. A 2-compartment pharmacokinetic model was implemented in the NONMEN VI program to determine the appropriate pharmacokinetic parameters. The power to identify the parameters was assessed by parametric bootstrap, and the internal model validation was performed using nonparametric bootstrap and visual and numerical predictive check methods. The final predictive performance of the model was assessed for accuracy and precision during the first (a priori) and second (a posteriori) chemotherapy cycles.
ResultsThe mean and interpatient variability of gemcitabine and dFdU clearance was 2.70L/min (31.0%) and 0.0515L/min (35.8%), respectively. The estimated distribution volume at steady state was 30L for gemcitabine and 238L for dFdU. Internal validation confirmed that the population pharmacokinetic model was appropriate for describing the plasma concentrations of gemcitabine and dFdU over time, as well as its variability in the study population. The accuracy and precision of a posteriori gemcitabine plasma concentrations improved by 67% and 46%, respectively, compared to the a priori prediction.
ConclusionThe population pharmacokinetic model adequately characterised the gemcitabine and dFdU plasma concentrations in the study population over time, and can be used to accurately and precisely optimise gemcitabine dosing regimens in cancer patients.
Gemcitabina es un antimetabolito análogo del arabinósido de citosina (Ara-C) cuya activación intracelular se produce tras su transporte al interior celular mediado por 5 transportadores de nucleósidos, ENT1, ENT2, CNT1, CNT2 y CNT31. Una vez dentro de la célula, la enzima deoxicitidina cinasa (dCK) fosforila la molécula de gemcitabina a gemcitabina monofosfato (dFdCMP). La fosforilación secuencial produce los metabolitos activos, gemcitabina difosfato (dFdCDP) y trifosfato (dFdCTP). El metabolito dFdCTP compite con deoxicitidina trifosfato en la unión a la ADN polimerasa2 y provoca la inhibición de la enzima, la terminación de la cadena de elongación del ADN e induce la apoptosis2. Además, se han descrito otras actividades citotóxicas como la inhibición de la síntesis de ARN dosis y tiempo dependiente3 y la inhibición de varias enzimas, entre las que destacan la enzima ribonucleótido reductasa4, la citidina trifosfato sintetasa5, la deoxicitidilato desaminasa6 y la topoisomerasa I7, enzimas que tienen un papel primordial en la síntesis y reparación del ADN.
Gemcitabina es un antineoplásico utilizado en diversos tipos de tumores sólidos, como el carcinoma de pulmón no microcítico localmente avanzado o metastásico, adenocarcinoma pancreático localmente avanzado o metastásico, cáncer de vejiga localmente avanzado o metastásico, carcinoma epitelial de ovario y adenocarcinoma de mama. Gemcitabina se administra habitualmente en perfusión intravenosa continua de 30 min de duración a una dosis inicial recomendada de 800-1.250mg/m2 semanal o cada 2 semanas, y puede administrarse en monoterapia o en combinación con cisplatino, carboplatino o paclitaxel.
Tras la administración intravenosa, gemcitabina se distribuye ampliamente en plasma y en distintos tejidos, y prácticamente no se une a las proteínas plasmáticas. No se ha descrito si atraviesa la barrera hematoencefálica, pero sí que se distribuye a la leche materna, circunstancia que justifica la interrupción de la lactancia en caso de administración de gemcitabina. El volumen de distribución en estado de equilibrio estacionario es de 17l/m2 (rango de 9 a 30l/m2) y el área bajo la curva de concentraciones plasmáticas frente al tiempo (AUC) es proporcional a dosis comprendidas entre 87 y 2.500mg/m2. La semivida biológica de gemcitabina oscila entre 42 y 94min, y su eliminación es completa tras un periodo de tiempo comprendido entre 5 y 11 h a partir del comienzo de la perfusión8. El aclaramiento sistémico oscila entre 29 y 92l/h/m2 dependiendo del género (variabilidad interindividual del 52%). En general, las mujeres presentan aclaramientos aproximadamente un 25% más bajos que los hombres8. Durante la semana posterior a la administración, entre el 92 y el 98% de la dosis de gemcitabina administrada se recupera mayoritariamente en la orina (99%), principalmente en la forma de 2’,2’- difluorodeoxiuridina (dFdU) y el resto de la dosis administrada se excreta por las heces8.
El metabolito dFdCTP se sintetiza en el interior de las células mononucleares sanguíneas periféricas y tiene una semivida biológica de 0,7 a 12h8. Por otra parte, gemcitabina es metabolizada a dFdU por la enzima citidina desaminasa (CDA) presente en hígado, riñones, sangre y otros tejidos. Esta enzima puede ser inhibida mediante la tetrahidrouridina, y por ello se utiliza para impedir ex vivo el paso de gemcitabina a su metabolito9. La dFdU tiene un volumen de distribución en estado equilibrio estacionario de 150 l/m2 (rango: 96 a 228l/m2). El aclaramiento aparente de dFdU es 2,5l/h/m2 (rango: 1 a 4l/h/m2) y su semivida biológica de la fase terminal es de, aproximadamente, 65h8. Se ha observado que la edad y el incremento de los niveles de creatinina sérica se correlacionan con un descenso del aclaramiento de dFdU10.
Los efectos adversos limitantes de dosis de gemcitabina son la neutropenia y trombocitopenia. La frecuencia de aparición y la severidad de las toxicidades hemáticas son mayores cuando se administra gemcitabina en combinación con paclitaxel, cisplatino o carboplatino8. Otras reacciones adversas asociadas al tratamiento con gemcitabina son náuseas, con o sin vómitos, aumento de las transaminasas hepáticas (aspartato aminotransferasa [AST], alanino aminotransferasa [ALT] y fosfatasa alcalina), proteinuria, hematuria, disnea y exantemas alérgicos. La frecuencia y la severidad de aparición de las reacciones adversas se ven influidas por la dosis, la velocidad de perfusión, los intervalos entre dosis y la administración de ciclos previos11.
Tras la administración de 1.000-1.250mg/m2 de gemcitabina en perfusión intravenosa continua de 30 min de duración se alcanzan concentraciones plasmáticas de gemcitabina de 15-20μM, y la formación de dFdCTP en las células mononucleares de sangre periférica se satura12. Así, la administración de dosis superiores a 1.500mg/m2 produce Cmáx superiores a 20μM que no incrementan el efecto antineoplásico y van acompañadas de un incremento de la gravedad de la toxicidad hematológica13. Diversos ensayos clínicos en fase II observaron que al prolongar el tiempo de perfusión intravenosa de gemcitabina se producía una mayor acumulación de los metabolitos en las células mononucleares sanguíneas periféricas y, por tanto, se incrementaba la eficacia del tratamiento14. A partir de estos resultados se propuso la administración en perfusión intravenosa a velocidad constante de 10mg/m2/min de gemcitabina como una alternativa a la perfusión intravenosa de 30 min14. Sin embargo, en el estudio realizado por Cappuzzo et al. se observó que la administración de gemcitabina a una perfusión a velocidad constante de 10mg/m2/min respecto a la administración en perfusión de 30 min no mejoraba significativamente la supervivencia media (9,8 meses en ambos grupos) ni el tiempo medio de progresión de la enfermedad (4,5 meses frente a 4 meses, p=0,34)15. Además, la perfusión a velocidad constante de 10 mg/m2/min va asociada a una mayor toxicidad hematológica con respecto a la perfusión de 30 min (neutropenia grado III-IV: 49,2 frente a 17,9%, p=0,0002)15. Por todo ello, en la práctica clínica habitual se utiliza más la perfusión intravenosa de 30 min que la perfusión constante de 10mg/m2/min.
La determinación de las concentraciones plasmáticas de gemcitabina y el potencial beneficio del ajuste de dosis en base a los parámetros farmacocinéticos se justifica principalmente por 3 razones: 1) es un fármaco con un margen terapéutico estrecho12,13; 2) presenta una elevada variabilidad en su farmacocinética10,16,17; 3) existe correlación entre sus concentraciones en plasma y su eficacia18 y toxicidad13. Las características farmacocinéticas y farmacodinámicas de gemcitabina justifican la monitorización de sus concentraciones plasmáticas aunque su implementación sistemática en la práctica clínica es escasa debido a la necesidad de disponer de una técnica analítica de adecuada precisión, exactitud, especificidad y selectividad para la determinación de gemcitabina en muestras biológicas de pacientes.
En cuanto a la individualización posológica, la metodología bayesiana es una de las técnicas más empleadas para la estimación de los parámetros farmacocinéticos individuales. La aplicación de esta técnica con garantía de éxito requiere la estimación exacta y precisa de los parámetros farmacocinéticos de gemcitabina y dFdU a partir de los datos correspondientes a la población específica de pacientes subsidiarios de ajuste posológico19. La farmacocinética de gemcitabina se ha descrito en la literatura médica mediante modelos farmacocinéticos lineales basados en dos compartimentos10,16-18, mientras que la farmacocinética de dFdU se ha descrito mediante modelos de uno17 o dos10,16 compartimentos. También se ha utilizado el modelo de efectos mixtos para caracterizar tanto la tendencia central como la variabilidad inter e intraindividual en los parámetros cinéticos de gemcitabina y dFdU, así como sus relaciones con covariables demográficas y clínicas10,16,17.
Por tanto, el objetivo del presente trabajo ha sido desarrollar y validar internamente un modelo farmacocinético poblacional para gemcitabina y dFdU y evaluar su capacidad predictiva en la personalización de su dosificación en pacientes oncológicos.
MétodosCriterios de selección de pacientesSe incluyeron en el estudio pacientes oncológicos adultos subsidiarios de recibir tratamiento con gemcitabina, con edad comprendida entre 18 y 80 años, con una expectativa de vida superior a 3 meses, función medular normal (recuento de leucocitos > 4 × 109/l, recuento de neutrófilos > 1,5 × 109/l y recuento de plaquetas > 150 × 109/l), función renal normal (creatinina sérica < 1,5mg/dl) y una función hepática conservada (bilirrubina < 1,2mg/dl, AST y ALT ≤ 40 U/l). Se excluyeron del estudio las mujeres lactantes o en periodo gestacional y aquellos pacientes que presentasen alguna de las siguientes situaciones: estado general mayor de 2 según la escala ECOG, o inferior a 60% en la escala Karnofsky, radioterapia extensa previa, trastornos psiquiátricos o de cualquier otro tipo que comprometiese la capacidad para otorgar el consentimiento informado verdadero para la participación en el estudio, cualquier circunstancia que impidiese que el tratamiento y su seguimiento se realizase según el protocolo establecido, como puede ser la progresión de enfermedad y haber participado en un estudio clínico al menos 30 días antes.
El estudio se llevó a cabo según los principios de investigación humana establecidos en la Declaración de Helsinki. En todos los casos se obtuvo el consentimiento informado del paciente tras una entrevista personal donde se le informó verbalmente y por escrito de los potenciales riesgos y beneficios del tratamiento y el estudio.
Toma de muestras y técnica analíticaEn los 2 primeros ciclos de quimioterapia se tomaron muestras sanguíneas a las 0,5, 0,75, 1,5 y 2,5 h desde el inicio de la perfusión. Estos tiempos fueron seleccionados en función de la teoría de muestreo óptimo y los modelos farmacocinéticos previamente publicados en la literatura biomédica. Las muestras de sangre se extrajeron del brazo contralateral al que se administró la quimioterapia y se recogieron en tubos de plástico con heparina de litio como anticoagulante (BD Vacutainer®) a los que se añadió tetrahidrouridina en proporción 1:100 como estabilizador. Las muestras se centrifugaron durante 10 min a 3.500rpm y el plasma se congeló a −80°C hasta su valoración.
La determinación de concentraciones plasmáticas de gemcitabina y dFdU se realizó mediante cromatografía líquida de alta resolución acoplada a detección de fluorescencia según una técnica analítica publicada previamente20. Antes del bioanálisis, las muestras plasmáticas se sometieron a una extracción líquido-líquido (rendimiento > 85% para gemcitabina y > 86% para dFdU). El análisis cromatográfico de gemcitabina y dFdU se realizó bajo las mismas condiciones de ensayo. Como fase estacionaria se empleó una columna cromatográfica Ultrabase C18 y una precolumna Ultrabase C18. Como fase móvil se empleó una mezcla de acetato amónico 0,01M (pH 5) y metanol en proporción 97:3 (v/v). La elución de los analitos fue isocrática y se realizó a un flujo de 1,7ml/min. La longitud de onda seleccionada en ambos casos fue de 258nm. La técnica analítica empleada permitió determinar concentraciones plasmáticas de gemcitabina entre 0,5 y 15μg/l con adecuada precisión (CV < 4%) y exactitud (error relativo < 8%). También se determinaron concentraciones plasmáticas de dFdU entre 1 y 70μg/l con adecuada precisión (CV< 2%) y exactitud (error relativo < 5%). El límite de cuantificación (LOQ) se determinó en 0,5μg/l y 1μg/l para gemcitabina y dFdU, respectivamente. La precisión y exactitud para el LOQ fue inferior al 20% en ambos casos.
Análisis farmacocinéticoProgramas informáticosEl análisis farmacocinético se realizó en el programa informático NONMEM VI versión 2.0 (ICON, Hanover, MD, USA)21 con la aproximación de LAPLACIAN22. La compilación del programa se realizó con el programa DIGITAL Visual Fortran versión 6.6C. Los gráficos y el resto de análisis estadísticos, incluyendo la evaluación de los modelos ajustados con NONMEM, se realizaron con el programa S-Plus 6.1 Professional Edition para Windows (Insightful, Seattle, WA, USA).
Modelo farmacoestadísticoTras el análisis exploratorio gráfico de los datos obtenidos y la evaluación de la literatura científica, se seleccionó un modelo farmacocinético bicompartimental, con distribución y eliminación lineal, tanto para gemcitabina como para dFdU. En este modelo se asume que la totalidad de la dosis administrada de gemcitabina se metaboliza a dFdU mediante un proceso lineal. Este modelo se parametrizó en términos de aclaramiento de gemcitabina (CLGEM, l/h), volumen de distribución central de gemcitabina (V1, l), flujo entre el compartimento central y el periférico de gemcitabina (Q2, l/h), volumen del compartimento periférico de gemcitabina (V2, l), aclaramiento de dFdU (CLDFDU, l/h), volumen de distribución central de dFdU (V3, l), flujo entre el compartimento central y el periférico liviano de dFdU (Q4, l/h) y volumen del compartimento periférico de dFdU (V4, l).
Se asumió que la variabilidad interindividual seguía una distribución log-normal según la ecuación:
donde P es el parámetro farmacocinético individual para el paciente j-ésimo, P* es el valor típico poblacional, (j es la variable independiente y aleatoria que cuantifica las diferencias entre el valor típico poblacional y su valor individual, y se asume que siguen una distribución normal de media cero y varianza ωP2. Las magnitudes de las variabilidades interindividuales determinadas en los parámetros de gemcitabina y dFdU representan aproximadamente los coeficientes de variación (CV). La variabilidad residual se evaluó utilizando un modelo de error aditivo tras la transformación logarítmica de las concentraciones observadas y las correspondientes predicciones del modelo, de acuerdo con la ecuación:donde Cobs es la concentración observada de gemcitabina o de dFdU en plasma, Cpred es la concentración predicha por el modelo, GD es una variable dummy que toma el valor 1 para las concentraciones de gemcitabina y el valor 0 para las concentraciones de dFdU, ¿gemcitabina y ¿dFdU son variables aleatorias independientes con distribución normal con media cero y varianza σgemcitabina2 y σdFdU2 para gemcitabina y dFdU, respectivamente, y describen la diferencia del logaritmo de la concentración observada y el logaritmo de la predicción del modelo, es decir, el valor residual.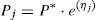
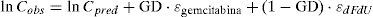
Además y dado que el número de concentraciones de gemcitabina por debajo del LOQ en este estudio fue superior al 10%, el modelo farmacoestadístico incluyó una función de probabilidad que permitió incluir esos datos en el análisis farmacocinético como datos censurados. Estos datos censurados se analizaron considerando que la probabilidad de una observación por debajo del LOQ se obtiene tras integrar la función de densidad log-normal desde menos infinito hasta el LOQ23. Este método que maximiza la probabilidad de todos los datos, reduce significativamente el sesgo en la estimación de los parámetros24.
Análisis de datosEl análisis famacocinético de las concentraciones plasmáticas de gemcitabina y dFdU se realizó mediante 4 métodos distintos. El método A se utilizó para estimar los parámetros farmacocinéticos poblacionales del modelo descrito por Jiang et al.16 a partir de las concentraciones de gemcitabina y dFdU obtenidas en la muestra de pacientes estudiada. Este método, denominado frecuentista, se utiliza en la literatura científica para caracterizar los parámetros de los modelos farmacocinéticos de gemcitabina y dFdU16, y necesita un número de muestras por paciente elevado, circunstancia que habitualmente no es factible en la práctica clínica rutinaria, como sucedió en este caso. Por tanto, el muestreo reducido empleado en el presente estudio podría resultar insuficiente para estimar con exactitud y precisión todos los parámetros del modelo farmacocinético poblacional. Ante esta situación, una alternativa (método B) fue el análisis bayesiano poblacional. Este método se utilizó para actualizar los parámetros farmacocinéticos poblacionales del modelo de Jiang et al. a partir de las concentraciones de gemcitabina y dFdU obtenidas en la muestra de pacientes estudiada. En este caso, los parámetros de efecto fijo y aleatorio establecidos por Jiang et al. se utilizaron como información a priori para la estimación bayesiana poblacional implementada en NONMEM mediante la subrutina $PRIOR con la opción NWPRI. Esta subrutina implementa una función de penalización derivada de los datos a priori mediante una distribución normal para los parámetros de efecto fijo y una distribución de Wishart invertida para los efectos aleatorios. Este método no incluye la información a priori para el error residual. Además, se definió la incertidumbre a priori de los parámetros de efecto fijo y los grados de libertad de los efectos aleatorios tal y como describen Gisleskog et al.25. Este método permite evaluar la capacidad para identificar los parámetros del modelo a partir de la información contenida en la base de datos analizada y la información disponible a priori. Así, cuando el error estándar de estimación de ciertos parámetros tiende a 0 se puede considerar que la base de datos contiene escasa información para caracterizar adecuadamente ciertos parámetros del modelo. Esta información puede utilizarse para decidir qué parámetros se deben fijar a un valor de la literatura puesto que la base de datos disponible no permite su estimación. Así, tras obtener las estimaciones por el método B, se fijaron aquellos parámetros cuyos errores estándar relativos (RSE) determinados con el método B fuesen igual a 0. Para estimar el resto de parámetros del modelos se plantearon 2 estrategias: una estimación frecuentista (método C) y una estimación bayesiana poblacional (método D).
Con objeto de evaluar el efecto que la estrategia de toma de muestras reducida tiene sobre la capacidad para identificar los parámetros del modelo farmacocinético poblacional de gemcitabina y dFdU se realizó un bootstrap paramétrico (PBT) de los métodos A, B, C y D. Así, se utilizaron los parámetros del modelo de Jiang et al. para generar 100 bases de datos diferentes aunque con idéntica estructura a la base de datos original. Cada una de las réplicas de la base de datos original, se analizó con los 4 métodos de análisis (A, B, C y D) y se obtuvieron los correspondientes parámetros farmacocinéticos poblacionales de tendencia central y variabilidad. A partir de estas estimaciones, se calculó la media, el error estándar y el intervalo de confianza del 95% de cada parámetro y se comparó con el valor del parámetro del modelo de Jiang et al. utilizado en la simulación de las bases de datos. Si el parámetro farmacocinético es identificable a partir de la base de datos disponible y el método de análisis utilizado, el intervalo de confianza del 95% de cada parámetro debería incluir el valor del parámetro obtenido por Jiang et al.
Validación del modeloLa validación interna del modelo se realizó mediante 3 técnicas diferentes y complementarias como son el bootstrap no paramétrico (NPBT), el visual predictive check (VPC) y el numerical predictive check (NPC). En el NPBT se consideró que el modelo no presentaba sesgos si las estimaciones de los parámetros del modelo desarrollado con la base de datos original se encontraban dentro del intervalo de confianza del 95% construido con bases de datos generadas mediante muestreo de pacientes con reemplazamiento con el programa WINGS para NONMEM VI (N. Holford, Version 616, Auckland, Nueva Zelanda). En el VPC, se compararon las concentraciones plasmáticas de gemcitabina y su metabolito correspondientes a la base de datos original con la media y el intervalo de predicción del 95% de concentraciones plasmáticas de gemcitabina y su metabolito simuladas a partir de la base de datos original y de los valores de los parámetros de efecto fijo y aleatorio estimados por el modelo final. Por último, en el NPC a partir de bases de datos simuladas con el modelo farmacocinético seleccionado, se estratificaron las concentraciones plasmáticas en 4 grupos en función del tiempo de muestreo (0,5, 0,75, 1,5 y 2,5 h desde el inicio de la perfusión). En cada grupo se calculó el valor medio y el CV (%) de las concentraciones plasmáticas de gemcitabina y su metabolito, así como los respectivos intervalos de confianza del 95% a partir de los percentiles 2,5, 50 y 97,5%. Estos resultados se compararon con la media y el CV (%) de las concentraciones observadas para cada tiempo.
Capacidad predictiva del modeloLa capacidad predictiva del modelo se evaluó en términos de exactitud y precisión de las concentraciones plasmáticas de gemcitabina y su metabolito dFdU según la teoría de errores de predicción de Sheiner y Beal26. Como parámetro de exactitud se calculó la media del error relativo medio de predicción (ERM) existente entre las concentraciones plasmáticas observadas y las correspondientes predicciones del modelo. Como parámetro de precisión se calculó la raíz cuadrada del error cuadrático medio de predicción (RECM). Los valores de ERM y RECM del modelo se calcularon mediante las ecuaciones 3 y 4, respectivamente:
donde Qi representa la estimación de la concentración plasmática obtenida en el i-ésimo paciente y Q* representa el valor de la concentración plasmática obtenido en ese mismo paciente. Además, se comprobó si el ERM difería significativamente (p<0,05) del valor de cero a través del cálculo de los intervalos de confianza del 95% de dicho valor.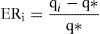
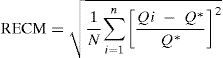
El análisis de la exactitud y la precisión se realizó por separado para gemcitabina y su metabolito dFdU, y para los 2 primeros ciclos de tratamiento. Las concentraciones plasmáticas predichas por el modelo para el primer ciclo de quimioterapia se obtuvieron en función de los parámetros típicos poblacionales (predicción a priori) puesto que no se disponía de información farmacocinética previa del paciente. Las concentraciones plasmáticas predichas por el modelo para el segundo ciclo de quimioterapia se generaron en función de los parámetros farmacocinéticos individuales del paciente (predicción a posteriori) obtenidos mediante estimación bayesiana individual a partir de las concentraciones plasmáticas observadas en el primer ciclo y el modelo poblacional desarrollado. De esta forma es posible evaluar el efecto de la monitorización de las concentraciones plasmáticas de gemcitabina y su metabolito dFdU en la optimización farmacoterapéutica de gemcitabina en pacientes oncológicos.
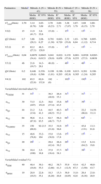
ResultadosEl presente estudio se ha realizado con un total de 18 pacientes oncológicos que recibieron tratamiento con gemcitabina intravenosa en un régimen de administración bisemanal, con una dosis media de 800mg/m2 (400-1.000mg/m2) y con una duración de la perfusión de 30 min. El tumor primario fue páncreas (n = 4), carcinoma vesical (n = 3), carcinoma de pulmón (n = 3), carcinoma de mama (n = 2), carcinoma de útero (n = 1), carcinoma de próstata (n = 1) y carcinoma de origen desconocido (n = 4). Según el tipo de tumor primario a tratar, la gemcitabina se administró en terapia combinada con otros antineoplásicos entre los que se incluyeron 5-fluorouracilo (28%), oxaliplatino (22%), docetaxel (17%), irinotecan (17%), paclitaxel (17%), carboplatino (11%) o doxorrubicina (11%), entre otros. Se monitorizaron un total de 30 ciclos de quimioterapia que supuso la cuantificación de la concentración plasmática de gemcitabina y dFdU en 83 y 111 muestras, respectivamente. En la tabla 1 se resumen las características de la población al inicio del estudio.
Resumen de las características basales de los pacientes y su tratamiento con gemcitabina
| Características de los pacientes (N=18) | Media (DE) | Ámbito |
| Edad (años) | 62 (11) | 46-84 |
| Superficie corporal (m2) | 1,8 (0,2) | 1,4-2,3 |
| Sexo (%) | ||
| Varones | 44,4 | - |
| Mujeres | 55,6 | - |
| ALT (IU/l) | 33,6 (8,7) | 19,0-40,0 |
| AST (IU/l) | 23,3 (6,8) | 12,0-37,0 |
| Fosfatasa alcalina (IU/l) | 210 (111) | 79-487 |
| Creatinina Sérica (mg/dl) | 0,8 (0,2) | 0,5-1,3 |
| Bilirrubina Total (mg/dl) | 0,4 (0,2) | 0,2-0,9 |
| Leucocitos × 109/l | 7,5 (3,4) | 4,1-14,4 |
| Neutrofilos × 109/l | 8,9 (18,3) | 1,5-8,1 |
| Plaquetas × 109/l | 307 (183) | 155-981 |
| Dosis de gemcitabina (mg) | ||
| Primer ciclo | 1408 (244) | 748-1800 |
| Segundo ciclo | 1361 (263) | 900-1800 |
| Concentraciones de gemcitabina (μg/l) | ||
| Primer ciclo (N=18) | ||
| 0,5 h | 9,6 (4,1) | 1,1-17,4 |
| 0,75 h | 2,5 (1,4) | 0,8-6,1 |
| 1,5 h | 0,4 (0,3) | 0,1-0,9 |
| 2,5 h | 0,5 (0,1) | 0,4-0,6 |
| Segundo ciclo (N=12) | ||
| 0,5 h | 8,7 (2,2) | 5,8-13,4 |
| 0,75 h | 3,3 (3,4) | 0,3-12,1 |
| 1,5 h | 0,4 (0,4) | 0,1-1,3 |
| 2,5 h | 0,4 (0,3) | 0,1-0,7 |
| Concentraciones de dFdU (μg/l) | ||
| Primer ciclo (N = 18) | ||
| 0,5 h | 20,6 (6,9) | 11,8-34,9 |
| 0,75 h | 27,2 (8,4) | 7,8-42,5 |
| 1,5 h | 21,2 (6,1) | 6,9-32,5 |
| 2,5 h | 15,6 (5,0) | 5,1-24,9 |
| Segundo ciclo (N =12) | ||
| 0,5 h | 14,4 (4,5) | 9,8-22,2 |
| 0,75 h | 22,3 (5,6) | 11,1-28,7 |
| 1,5 h | 16,1 (3,9) | 7,9-20,2 |
| 2,5 h | 13,2 (3,2) | 6,6-18,4 |
ALT: alanina aminotransferasa; AST: aspartato aminotransferasa; DE: desviación estándar.
Nota. Las variables continuas están expresadas como media (DE) mientras que las variables categóricas están expresadas en porcentaje (%).
Los resultados del análisis farmacocinético se muestran en la tabla 3. Estos resultados no pueden interpretarse de manera aislada respecto a los resultados del PBT realizado con los métodos A, B, C y D para evaluar el efecto que la estrategia de toma de muestras reducida tiene sobre la capacidad para identificar los parámetros del modelo farmacocinético poblacional de gemcitabina y dFdU y que se recogen en la tabla 2. Con el método A la estimación de los parámetros farmacocinéticos de efecto fijo tanto para gemcitabina como para dFdU se modificaron entre un 30 y un 330% respecto a los valores referidos en la literatura (tabla 2)16. Estos resultados evidencian la imposibilidad de identificar correctamente los parámetros del modelo a partir de los datos experimentales. La estrategia basada en el análisis bayesiano poblacional (método B) permitió identificar tanto los parámetros de efecto fijo como los de efecto aleatorio. No obstante, los RSE de los volúmenes centrales y periféricos de gemcitabina y dFdU fueron 0, circunstancia que justifica la evaluación del método C y D como alternativa a los métodos A y B. Por tanto, el valor de estos parámetros se fijó en el método C y D, al valor estimado por Jiang et al. Al fijar los volúmenes centrales y periféricos de gemcitabina y dFdU a los valores referidos en la literatura especializada (método C), fue necesario fijar además el valor de las variabilidades interindividuales para que el modelo minimizara satisfactoriamente. Las estimaciones del resto de parámetros difieren más de un 20% respecto a los valores determinados por Jiang et al. y sus correspondientes RSE aumentaron hasta un 200%. Además, al fijar 4 parámetros de efecto fijo y 8 de efecto aleatorio se perdió capacidad predictiva por cuanto que los errores residuales de gemcitabina y dFdU aumentaron hasta un 45% y un 97,5% con el método C, respectivamente, respecto a los parámetros de Jiang et al. Con el método D, tanto los parámetros de efecto fijo como los de efecto aleatorio son perfectamente identificables con aceptables errores estándar de estimación. Estos resultados permiten explicar las diferencias en los parámetros determinados con los 4 métodos y seleccionar el modelo D como primera elección para analizar los datos experimentales disponibles.
Parámetros farmacocinéticos poblacionales y análisis bootstrap paramétrico realizado con los métodos A, B, C y D
| Parámetros | Mediaa | Método A (N=43) | Método B (N=55) | Método C (N=62) | Método D (N=36) | ||||
| Media (RSE) | IC 95% | Media (RSE) | IC 95% | Media (RSE) | IC 95% | Media (RSE) | IC 95% | ||
| ClGEM(l/min) | 2,70 | 3,14 (9,78) | 2,63-3,69 | 2,70 (0,23) | 2,69-2,71 | 3,28 (7,87) | 2,87-3,79 | 2,69 (0,15) | 2,69-2,70 |
| V1(l) | 15 | 11,9 (41,7) | 3,8-22,6 | 15 (0) | – | 15b | – | 15b | – |
| Q2 (l/min) | 0,7 | 3,66 (29,3) | 2,08-6,27 | 0,701 (1,30) | 0,683-0,720 | 2,32 (53,6) | 1,03-6,04 | 0,706 (1,12) | 0,695-0,714 |
| V2 (l) | 15 | 82,2 (27,3) | 48,3-120,9 | 15 (0) | – | 15b | – | 15b | – |
| ClDFDU(l/min) | 0,04 | 0,0079 (34,4) | 0,0045-0,0121 | 0,041 (28,9) | 0,022-0,058 | 0,139 (57,6) | 0,001-0,253 | 0,0528 (27,5) | 0,0283-0,0638 |
| V3 (l) | 46 | 21,6 (19,7) | 14,3-29,1 | 46 (0) | – | 46b | – | 46b | – |
| Q4 (l/min) | 0,2 | 0,446 (16,8) | 0,339-0,586 | 0,199 (1,01) | 0,196-0,203 | 0,109 (92,8) | 0,013-0,305 | 0,201 (1,24) | 0,198-0,205 |
| V4 (l) | 192 | 85,5 (22,9) | 62,0-132,8 | 192 (0) | – | 192b | – | 192b | – |
| Variabilidad interindividual (%) | |||||||||
| ωClGEM | 31 | 31b | – | 30,3 (40,9) | 26,4-35,1 | 31b | – | 31b | – |
| ωV1 | 39 | 73,5 (107) | 12,5-152,9 | 38,6 (37,4) | 35,8-43,5 | 39b | – | 39b | – |
| ωQ2 | 44 | 21,2 (134) | 3,4-41,3 | 44,5 (40,5) | 40,3-53,2 | 44b | – | 22,2 (86,8) | 14,19-33,11 |
| ωV2 | 64 | 56,6 (87,9) | 11,1-83,5 | 62,7 (46,7) | 56,2-73,5 | 64b | – | 64b | – |
| ωCLDFDU | 35 | 150 (69,9) | 92-213 | 35,5 (21,8) | 34,9-36,6 | 35b | – | 45,8 (131) | 13,3-91,6 |
| ωV3 | 15 | 48,6 (78,6) | 33,2-164 | 15,0 (38,9) | 13,6-17,8 | 15b | – | 15b | – |
| ωQ4 | 29 | 29b | – | 29,4 (42,4) | 26,4-36,2 | 29b | – | 13,5 (84,2) | 8,14-19,6 |
| ωV4 | 38 | 10,4 (108) | 3,5-18,8 | 37,9 (16,4) | 37,7-38,1 | 38b | 38b | – | |
| Variabilidad residual (%) | |||||||||
| σGEM | 40 | 48,4 (10,9) | 39,1-55,3 | 40,2 (2,68) | 38,7-41,3 | 55,9 (12,5) | 43,4-67,3 | 42,5 (3,50) | 40,0-43,7 |
| σDFDU | 19 | 28,9 (10,5) | 22,8-35,1 | 18,3 (8,20) | 15,3-20,8 | 36,9 (8,05) | 31,0-42,6 | 26,4 (8,16) | 23,8-28,9 |
IC: intervalo de confianza; RSE: error estándar relativo.
Los resultados del análisis farmacocinético de las concentraciones plasmáticas mediante el método D se muestran en la tabla 3 junto con los resultados del NPBT. Las estimaciones de los parámetros de efecto fijo y efecto aleatorio son similares a las obtenidas con el modelo de Jiang et al. Los correspondientes RSE de los efectos fijos son inferiores al 6 y al 36% para gemcitabina y dFdU, respectivamente. De las mil réplicas analizadas durante el NPBT, un 54,9% no minimizaron con éxito debido a errores de redondeo y se excluyeron del análisis. Las estimaciones poblacionales del modelo bayesiano fueron similares a la media de las réplicas generadas por el NPBT y estaban contenidas en el intervalo de confianza del 95%. Estos resultados demuestran la adecuada exactitud y precisión de los parámetros estimados mediante la metodología bayesiana poblacional.
Parámetros farmacocinéticos poblacionales y análisis bootstrap no paramétrico
| Parámetros | Método AMedia (RSE)a | Método BMedia (RSE)a | Método CMedia (RSE) | Método DMedia (RSE) | Boostrap no paramétrico‡ (método D) | |
| Media (RSE) | IC 95% | |||||
| ClGEM(l/min) | 3,32 | 2,70 | 3,14 (8,31) | 2,70 (1,16) | 2,70 (0,40) | 2,69-2,73 |
| 1 | ||||||
| V1(l) | 10,1 | 15 | 15b | 15b | 15b | – |
| Q2 (l/min) | 3,49 | 0,727 | 1,64 (18,8) | 0,728 (5,99) | 0,705 (3,18) | 0,649-0,733 |
| V2(l) | 64,9 | 15 | 15b | 15b | 15b | – |
| CLDFDU(l/min) | 0,00985 | 0,0599 | 0,114 (19,5) | 0,0548 (35,2) | 0,0515 (29,7) | 0,0308-0,0866 |
| V3 (l) | 23,7 | 46 | 46b | 46b | 46b | – |
| Q4 (l/min) | 0,454 | 0,204 | 0,133 (35,4) | 0,203 (6,69) | 0,200 (1,31) | 0,196-0,205 |
| V4 (l) | 76,1 | 192 | 192b | 192b | 192b | – |
| Variabilidad interindividual (%) | ||||||
| ωClGEM | 31 | 28,4 | 31b | 31b | 31b | – |
| ωV1 | 48,2 | 41,8 | 39b | 39,0b | 39b | – |
| ωQ2 | 18,6 | 42,1 | 44b | 41,1 (7,82) | 21,5 (75,8) | 13,4-33,8 |
| ωV2 | 62,2 | 63,9 | 64b | 64b | 64b | – |
| ωCLDFDU | 109 | 35,6 | 35b | 35,8 (58,0) | 28,1 (97,7) | 12,4-49,7 |
| ωV3 | 49,6 | 28,2 | 15b | 15,0b | 15b | – |
| ωQ4 | 29 | 29,1 | 29b | 28,6 (48,4) | 14,1 (91,1) | 7,0-23,7 |
| ωV4 | 9,53 | 37,8 | 38b | 38,0b | 38b | – |
| Variabilidad residual (%) | ||||||
| σGEM | 50,8 | 46,8 | 58,0 (14,0) | 46,8 (5,17) | 42,0 (4,29) | 39,3-45,8 |
| σDFDU | 29,8 | 28,7 | 37,0 (23,3) | 30,4 (5,49) | 26,7 (4,64) | 24,1-28,9 |
IC: intervalo de confianza; RSE: error estándar relativo.
En la figura 1 se presentan los resultados del VPC del método D, donde se evidencia que la evolución temporal de las concentraciones plasmáticas de gemcitabina y dFdU y su variabilidad es caracterizada de forma adecuada. De hecho, del total de concentraciones experimentales de gemcitabina y dFdU, un 4,0% (IC 95%: 0,1-7,9%) y un 6,3% (IC 95%: 1,8-10,8%) quedaron fuera del intervalo de predicción del 95%, respectivamente. Estos resultados confirman que el modelo describe adecuadamente los datos observados en la población de pacientes de estudio, tanto en el primer ciclo como en el segundo ciclo de tratamiento.
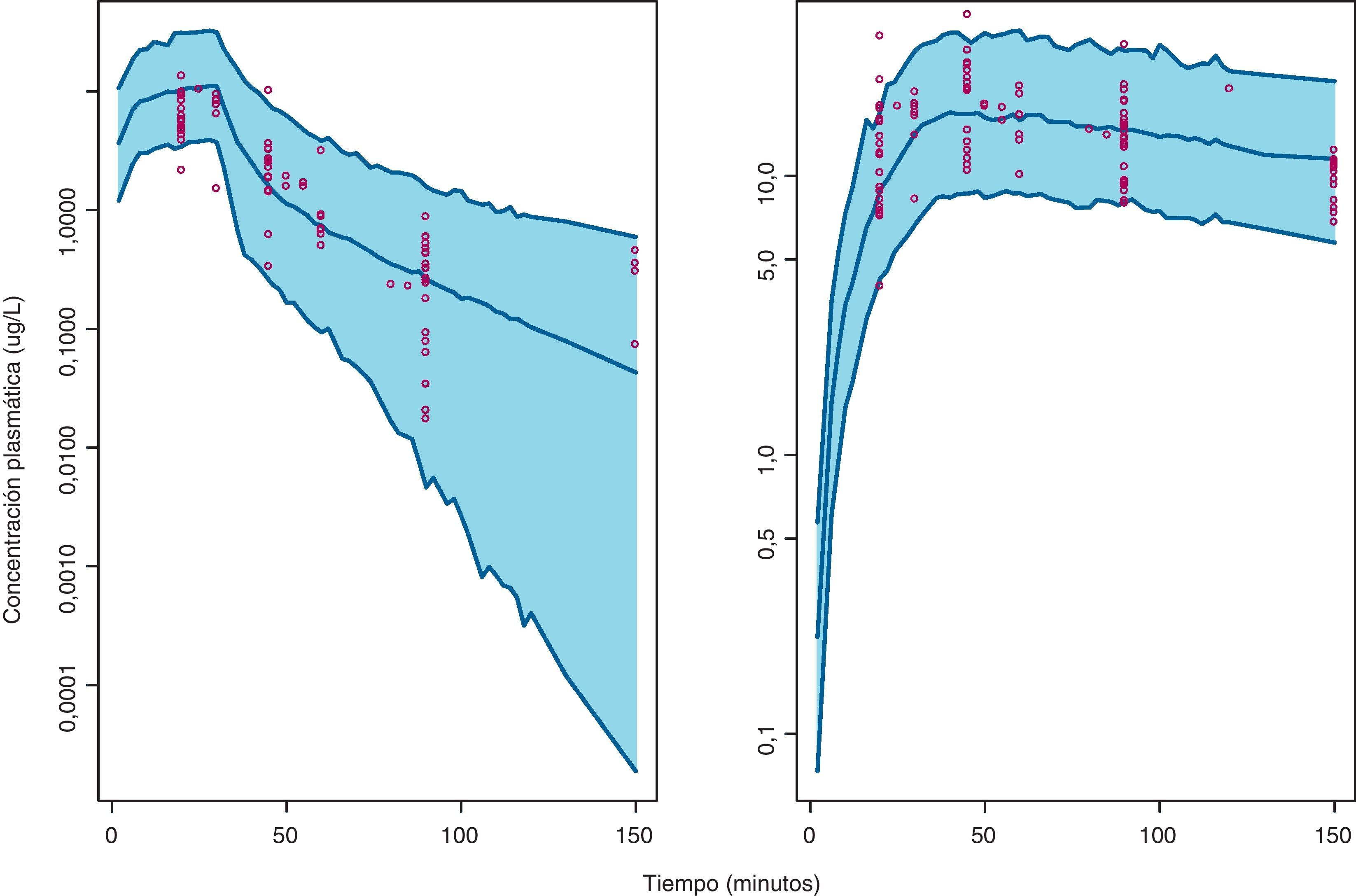
Visual Predictive Check de la evolución temporal de las concentraciones plasmáticas de gemcitabina (izquierda) y dFdU (derecha) en primer y segundo ciclo. En cada panel, las líneas azules continuas representan el límite superior, la media y el límite inferior, en la predicción de concentraciones al 95%, el área azul sombreada representa el intervalo de predicción del 95% y los puntos rojos representan las concentraciones de gemcitabina y dFdU observadas en plasma durante los primeros 2 ciclos de quimioterapia.
La figura 2 recoge los resultados del NPC (método D) para la gemcitabina y el dFdU en los dos parámetros evaluados en cada tiempo de muestreo: la concentración plasmática media y su correspondiente coeficiente de variación (CV%). En todos los paneles las barras del histograma representan la densidad de probabilidad de los datos simulados, la línea vertical punteada azul representa los percentiles 2,5, 50, y 97,5 de la distribución simulada y la línea vertical roja representa el valor del parámetro obtenido a partir de la base de datos original. Como se puede observar, las concentraciones medias en la población estudiada se incluyen dentro del intervalo de confianza en todos los casos, excepto en el caso de gemcitabina para el tiempo de 2,5 h donde el valor observado, 0,30μg/l fue ligeramente superior al del IC 95% estimado, 0,27μg/l. En cuanto al CV(%) en todos los casos el valor observado se encuentra dentro del intervalo de predicción simulado. Por tanto, estos resultados en su conjunto confirman la validez del método D para describir tanto las concentraciones plasmáticas de gemcitabina y dFdU como su variabilidad.
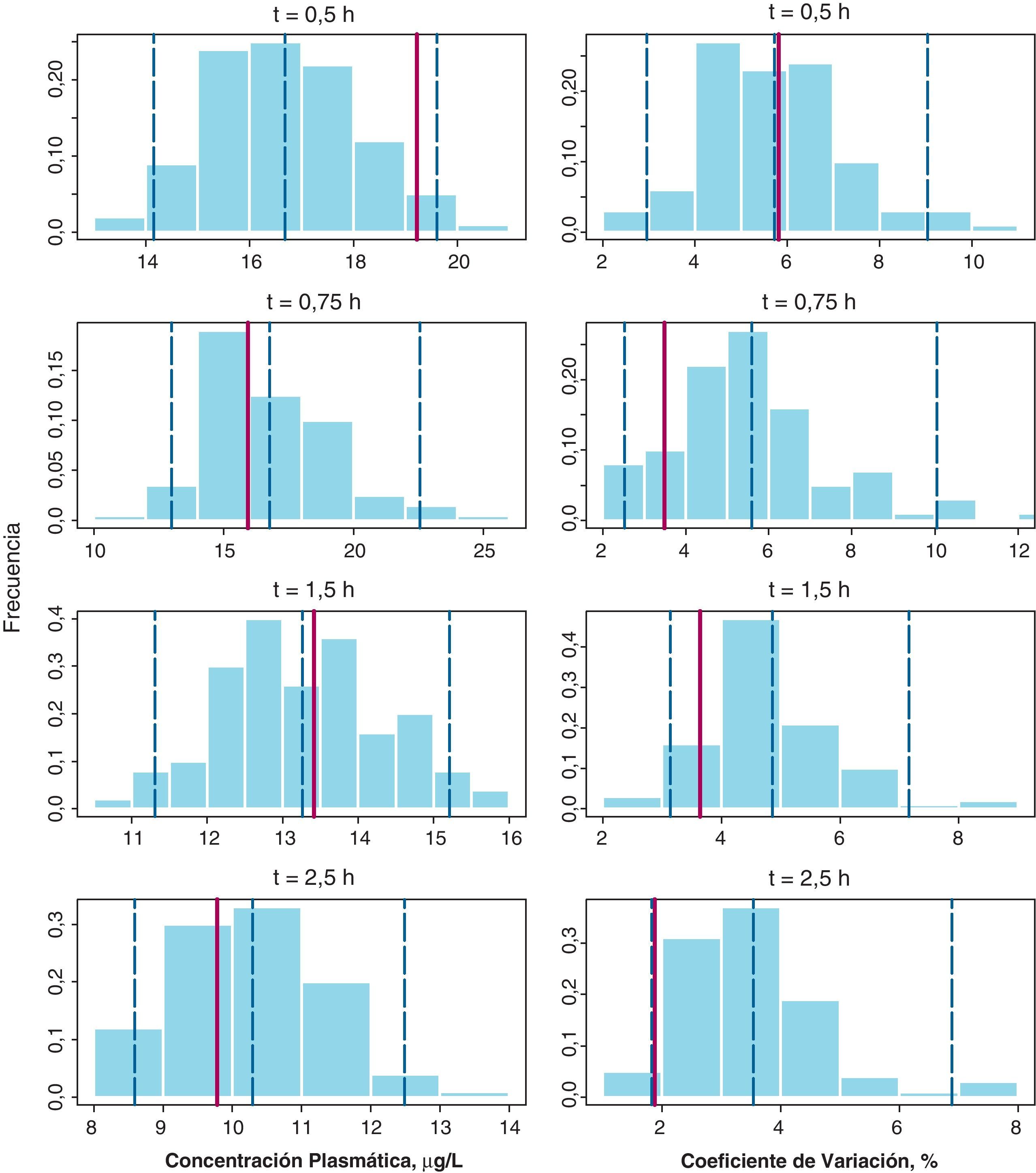
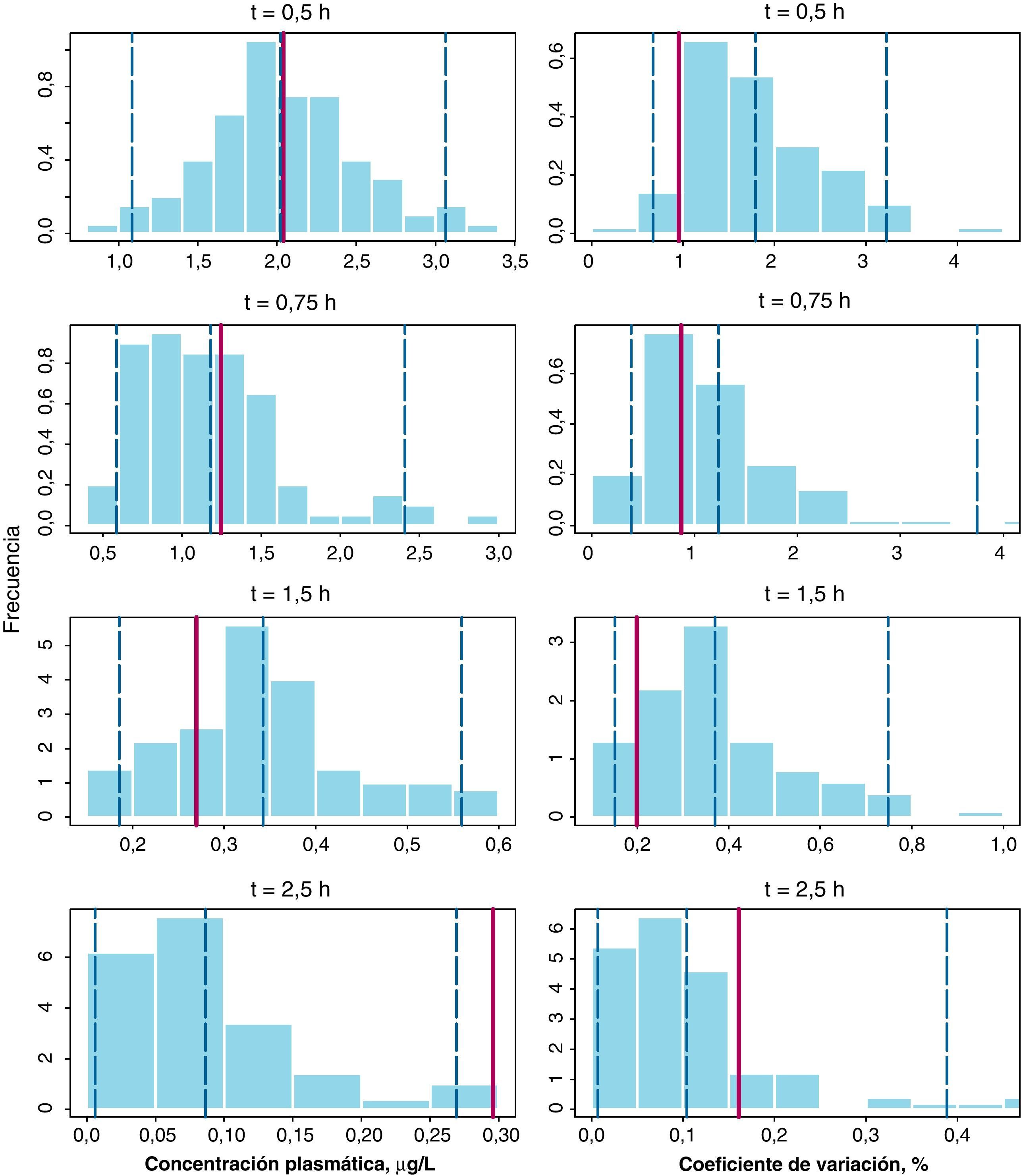
Numerical Predictive Check para la media de concentraciones plasmáticas de gemcitabina a cada uno de los tiempos de recogida de muestra (paneles de la izquierda) junto con sus coeficientes de variación (paneles de la derecha). Las líneas verticales discontinuas representan los percentiles 2,5, 50 y 97,5 de la distribución de concentraciones simuladas y la línea vertical roja continua representa la correspondiente observada en la muestra de paciente analizada, respectivamente.
Por último, los resultados de la evaluación de la capacidad predictiva del modelo seleccionado se resumen en la tabla 4. La predicción de las concentraciones plasmáticas de gemcitabina a priori presentó un sesgo sistemático ya que el IC 95% no incluyó el valor cero. La información individual del paciente, obtenida tras la monitorización de las concentraciones plasmáticas de gemcitabina en el primer ciclo de quimioterapia, mejoró estadísticamente en la capacidad predictiva del modelo por cuanto que el ERM mejoró un 67% y eliminó el sesgo sistemático en la predicción de las concentraciones plasmáticas de gemcitabina. Respecto al primer ciclo de quimioterapia, el RECM mejoró un 46% en el segundo ciclo, indicando una mejora en la precisión de la predicción de las concentraciones plasmáticas de gemcitabina. Para dFdU, la exactitud en el segundo ciclo no se redujo respecto a la estimada en el primer ciclo. Sin embargo, respecto al primer ciclo de quimioterapia, el RECM mejoró un 40% en el segundo ciclo.
Capacidad predictiva de gemcitabina y dFdU del modelo final en primer y segundo ciclo de quimioterapia
| Exactitud (%) | Precisión (%) | |||||
| Estimación | Bootstrap | Estimación | Bootstrap | |||
| Media | IC 95% | Media | IC 95% | |||
| Gemcitabina | ||||||
| Ciclo 1 | 34,4 | 34,2 | 6,7-87,8 | 125 | 129 | 66-222 |
| Ciclo 2 | 11,2 | 10,6 | −14,0-36,7 | 66,9 | 66,8 | 52,6-84,8 |
| dFdU | ||||||
| Ciclo 1 | -4,8 | -4,3 | −11,6-3,3 | 32,3 | 32,3 | 26,9-38,2 |
| Ciclo 2 | 4,1 | 4,3 | −6,4-15,0 | 19,4 | 19,6 | 12,6-31,5 |
IC: intervalo de confianza.
El primer objetivo de este trabajo ha sido desarrollar un modelo farmacocinético poblacional para caracterizar la evolución temporal de las concentraciones plasmáticas de gemcitabina y dFdU en pacientes oncológicos. Así, se ha determinado que la farmacocinética de gemcitabina y dFdU se describe adecuadamente mediante un modelo bicompartimental con distribución y eliminación de primer orden en ambos casos. La exploración inicial de los datos puso de manifiesto que un 18% de las concentraciones plasmáticas de gemcitabina estaban por debajo del LOQ. En el caso de dFdU, todas las concentraciones plasmáticas estaban por encima del LOQ. En la literatura científica se describen fundamentalmente tres métodos diferentes de análisis de las concentraciones por debajo del LOQ. El primer método se basa en la omisión de la base de datos de las concentraciones por debajo del LOQ. En un estudio realizado por Beal se evidenció que este método introduce sesgos relevantes en la estimación de los parámetros24. El segundo método se basa en la asignación de algún valor fijado relacionado con el LOQ, como por ejemplo fijarlo al valor de LOQ/2 o al valor cero24. Este método reduce el sesgo en la estimación de los parámetros respecto al método anterior. Sin embargo, el sesgo en la estimación sigue siendo relevante, especialmente cuando la proporción de concentraciones por debajo del LOQ respecto a las totales es elevada27. Por tanto, los dos métodos descritos deben evitarse, especialmente cuando los LOQ no sean completamente aleatorios. El tercer método se basa en maximizar la probabilidad de todos los datos, tratando las concentraciones por debajo del LOQ como datos censurados. Este método se ha utilizado en el presente trabajo y diversas publicaciones científicas han demostrado que reduce significativamente el sesgo en la estimación de los parámetros27.
Se ha seleccionado como modelo final el modelo bicompartimental tanto para gemcitabina como para dFdU, descrito por Jiang et al. Los parámetros farmacocinéticos se han estimado mediante la aproximación bayesiana poblacional y se han fijado previamente aquellos parámetros cuya limitada información impide una estimación adecuada del parámetro de interés. En este sentido, las estimaciones de los parámetros de efecto fijo y aleatorio han sido similares a las del modelo de Jiang et al. y los RSE de los parámetros de efecto fijo inferiores al 36%.
Los resultados de la validación interna del modelo farmacocinético seleccionado han confirmado que el modelo predice adecuadamente la evolución temporal de las concentraciones de gemcitabina y dFdU y su variabilidad y, por lo tanto, puede utilizarse para la optimización farmacoterapéutica de los tratamientos oncológicos con gemcitabina.
Aunque en la literatura científica se describe el efecto del género y la edad en la farmacocinética de gemcitabina, debido al escaso número de sujetos incluidos en el presente estudio, no se ha podido determinar los efectos de estas covariables en los parámetros farmacocinéticos, aspecto que se evaluará posteriormente cuando el número de pacientes monitorizados sea mayor y se disponga de suficiente poder estadístico para evaluar estas correlaciones. Por otra parte, sería interesante evaluar el efecto en la farmacocinética de gemcitabina de diversos polimorfismos de las principales enzimas implicadas en su metabolización, como por ejemplo la enzima citidina desaminasa28, así como otros biomarcadores responsables de la toxicidad hematológica de gemcitabina, como por ejemplo la haptoglobina29.
Para evaluar el efecto que la optimización farmacoterapéutica puede tener en la individualización de pautas posológicas de gemcitabina, se ha determinado la capacidad predictiva del modelo seleccionado en términos de exactitud y precisión. Por otra parte, no se determinaron los metabolitos activos dFdCDP y dFdCTP puesto que no se disponía de métodos analíticos para su determinación intracelular. Los resultados indican que en el primer ciclo el modelo presenta un sesgo del 34% en la predicción de las concentraciones plasmáticas de gemcitabina dado que tienen más peso las estimaciones de la población a priori respecto a la población de estudio. Sin embargo, en el segundo ciclo, al introducir los datos de concentraciones plasmáticas del paciente determinadas tras el primer ciclo de quimioterapia, el sesgo desaparece y la precisión aumenta un 46%. Respecto a dFdU, en el segundo ciclo no se mejora la exactitud respecto al primer ciclo, sin embargo, la precisión mejora un 40%.
En conclusión, el modelo farmacocinético poblacional caracteriza adecuadamente la evolución temporal de las concentraciones plasmáticas de gemcitabina y dFdU en la población de estudio y puede utilizarse de forma exacta y precisa para optimizar las pautas posológicas de gemcitabina en pacientes oncológicos.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
La Dra. Amelia Ramón López ha realizado el presente trabajo durante una estancia de investigación en la Unidad de Farmacoterapia Personalizada, en el amparo de un convenio de colaboración entre USP Hospital San Jaime y la Universidad Miguel Hernández.

