



El objetivo de las agencias reguladoras es asegurar un balance beneficio/riesgo favorable para los medicamentos en su indicación autorizada, sin entrar a establecer su lugar en la terapéutica más allá de eso. La indicación autorizada abarca subgrupos heterogéneos y a menudo no especifica lo suficiente las características de los pacientes que se pueden beneficiar. La información regulatoria no muestra siempre el beneficio frente al tratamiento o tratamientos estándar; además, se limita a responder exclusivamente a las condiciones especificadas en la solicitud del promotor y carece de valoración de la relevancia clínica del beneficio y sus incertidumbres.
Numerosos casos revelan la necesidad de establecer un escenario de utilidad terapéutica más específico que la indicación autorizada. Así, por ejemplo, abemaciclib se autorizó en adyuvancia para pacientes de alto riesgo con cáncer de mama precoz, pero es preciso especificar el nivel de riesgo adecuado y la forma de valorarlo. Igualmente, pembrolizumab está autorizado en neoadyuvancia más adyuvancia en cáncer de pulmón, pero falta analizar si es superior a nivolumab en neoadyuvancia solamente, que supone menor carga de tratamiento y económica.
Puesto que el posicionamiento terapéutico es una decisión siempre necesaria, ya se tome a nivel estatal, regional, local o individual, es preciso realizarlo de la forma más adecuada posible. Prescindir de una discusión y consenso multidisciplinares, pretendiendo que sean exclusivamente las decisiones individuales las que vayan estableciendo el posicionamiento desde el principio, implica subestimar las carencias de información, la variabilidad interindividual y la influencia de la promoción. En consecuencia, puede resultar perjudicial y oneroso.
Para gestionar de forma adecuada la introducción de nuevos medicamentos, resulta imprescindible establecer su escenario de utilidad de forma multidisciplinar. Esto, unido a considerar el beneficio clínico aportado frente a las alternativas adecuadas, así como las incertidumbres del mismo, constituye el objetivo de la evaluación clínica y la base para diseñar un análisis económico bien enfocado. Así, las autoridades pueden tomar las decisiones más adecuadas sobre el precio y financiación de los nuevos tratamientos. En una situación ideal, el escenario de utilidad considerado para el nuevo medicamento coincidiría con lo establecido para la financiación, pero costes difícilmente asumibles pueden conllevar restricciones y afectar al posicionamiento final, tras la evaluación económica y de impacto presupuestario.
The objective of regulatory authorities is to ensure a favorable risk–benefit balance for medicines in their licensed indication, without seeking to establish their place in the therapeutic armamentarium beyond that. The licensed indication covers heterogeneous subpopulations and often does not sufficiently specify the characteristics of the patients who may benefit. The regulatory information does not always show the benefit over the standard treatments; moreover, it only reacts to the conditions specified in the developer's application, and lacks an assessment of the clinical relevance of the benefit and its uncertainties.
Many cases highlight the need to establish a more specific therapeutic benefit scenario than the licensed indication. For example, abemaciclib was approved in the adjuvant setting for high-risk patients with early breast cancer, but the appropriate level of risk and how to assess it needs to be specified. Also, pembrolizumab is approved for neoadjuvant plus adjuvant treatment in lung cancer; but it remains to be analyzed whether it is superior to nivolumab in neoadjuvant treatment alone, which involves less treatment and economic burden.
As therapeutic positioning is always a necessary decision, whether made at a national, regional, local or individual level, it must be made in the most appropriate way. The absence of a multidisciplinary discussion and consensus, relying only on individual decisions to determine positioning from the outset, underestimates information gaps, inter-individual variability and the influence of drug promotion. It can be harmful and costly.
To properly manage the introduction of new medicines, it is essential to establish their benefit scenario in a multidisciplinary way. This, together with consideration of the clinical benefit provided versus the appropriate alternatives and the uncertainties of the benefit, constitutes the objective of the clinical assessment and the basis for designing a well-focused economic analysis. This allows policy makers to make the most appropriate decisions on pricing and funding new treatments. In an ideal situation, the benefit scenario considered for the new medicine would coincide with the one established for funding, but costs that are difficult to bear may lead to restrictions and affect the final positioning after the economic and budgetary impact assessment.
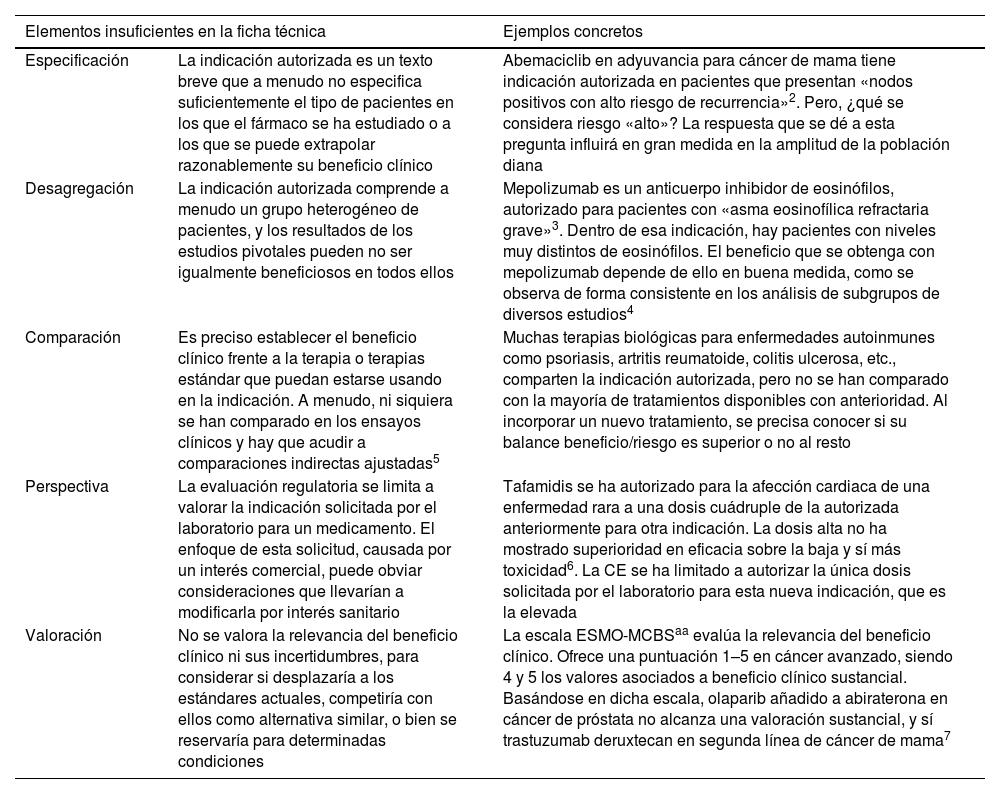
La evaluación realizada por las agencias regulatorias (como EMA y FDA) se está limitando al objetivo de asegurar que la relación beneficio/riesgo de los nuevos medicamentos sea positiva a nivel poblacional, de forma que estos puedan ser comercializados y prescritos valorando su utilidad potencial en los pacientes individuales1. Esta utilidad potencial se concreta en la ficha técnica, que contiene la indicación autorizada. Pero la ficha técnica no pretende responder a todas las cuestiones sobre qué lugar concreto puede ocupar un fármaco en la terapéutica real, en comparación con las demás opciones de tratamiento. Las carencias de la información regulatoria pública para guiar la introducción de un nuevo tratamiento en terapéutica –aparte de las muy importantes consideraciones económicas– se puede resumir en 5 aspectos esenciales (tabla 1).
Carencias de la información regulatoria pública para guiar la introducción de un nuevo tratamiento en la práctica clínica
| Elementos insuficientes en la ficha técnica | Ejemplos concretos | |
|---|---|---|
| Especificación | La indicación autorizada es un texto breve que a menudo no especifica suficientemente el tipo de pacientes en los que el fármaco se ha estudiado o a los que se puede extrapolar razonablemente su beneficio clínico | Abemaciclib en adyuvancia para cáncer de mama tiene indicación autorizada en pacientes que presentan «nodos positivos con alto riesgo de recurrencia»2. Pero, ¿qué se considera riesgo «alto»? La respuesta que se dé a esta pregunta influirá en gran medida en la amplitud de la población diana |
| Desagregación | La indicación autorizada comprende a menudo un grupo heterogéneo de pacientes, y los resultados de los estudios pivotales pueden no ser igualmente beneficiosos en todos ellos | Mepolizumab es un anticuerpo inhibidor de eosinófilos, autorizado para pacientes con «asma eosinofílica refractaria grave»3. Dentro de esa indicación, hay pacientes con niveles muy distintos de eosinófilos. El beneficio que se obtenga con mepolizumab depende de ello en buena medida, como se observa de forma consistente en los análisis de subgrupos de diversos estudios4 |
| Comparación | Es preciso establecer el beneficio clínico frente a la terapia o terapias estándar que puedan estarse usando en la indicación. A menudo, ni siquiera se han comparado en los ensayos clínicos y hay que acudir a comparaciones indirectas ajustadas5 | Muchas terapias biológicas para enfermedades autoinmunes como psoriasis, artritis reumatoide, colitis ulcerosa, etc., comparten la indicación autorizada, pero no se han comparado con la mayoría de tratamientos disponibles con anterioridad. Al incorporar un nuevo tratamiento, se precisa conocer si su balance beneficio/riesgo es superior o no al resto |
| Perspectiva | La evaluación regulatoria se limita a valorar la indicación solicitada por el laboratorio para un medicamento. El enfoque de esta solicitud, causada por un interés comercial, puede obviar consideraciones que llevarían a modificarla por interés sanitario | Tafamidis se ha autorizado para la afección cardiaca de una enfermedad rara a una dosis cuádruple de la autorizada anteriormente para otra indicación. La dosis alta no ha mostrado superioridad en eficacia sobre la baja y sí más toxicidad6. La CE se ha limitado a autorizar la única dosis solicitada por el laboratorio para esta nueva indicación, que es la elevada |
| Valoración | No se valora la relevancia del beneficio clínico ni sus incertidumbres, para considerar si desplazaría a los estándares actuales, competiría con ellos como alternativa similar, o bien se reservaría para determinadas condiciones | La escala ESMO-MCBSaa evalúa la relevancia del beneficio clínico. Ofrece una puntuación 1–5 en cáncer avanzado, siendo 4 y 5 los valores asociados a beneficio clínico sustancial. Basándose en dicha escala, olaparib añadido a abiraterona en cáncer de próstata no alcanza una valoración sustancial, y sí trastuzumab deruxtecan en segunda línea de cáncer de mama7 |
La evaluación regulatoria concluye con una autorización de comercialización por la Comisión Europea (CE). A continuación, es precisa la solicitud de precio y financiación por parte del laboratorio en cada país, lo cual puede retrasarse. Tras una negociación con el financiador público, se emite una decisión de precio y financiación. Si es negativa, el laboratorio puede comercializarlo con un uso limitado al ámbito privado y al precio que decida, o bien puede abstenerse de lanzarlo al mercado.
Para el desarrollo de este proceso de negociación y decisión, es preciso concretar el lugar que va a ocupar el medicamento en terapéutica, así como una evaluación de los aspectos económicos. La información regulatoria y la decisión acerca de la indicación autorizada aportan información fundamental, pero es preciso añadir una evaluación comparativa y tomar decisiones adicionales, con el fin de obtener el mayor beneficio para los pacientes de forma eficiente.
Gestión de una introducción razonable en terapéuticaToda nueva terapia que se introduce en la práctica clínica va a ocupar un nicho o lugar en terapéutica y, a menos que cubra una situación antes, sin tratamiento o «laguna terapéutica», acabará desplazando a otros tratamientos, bien por ser preferible para la salud, comodidad de los pacientes o facilidad de su aplicación, o bien por ser una alternativa similar que compita en costes. Esta fase de posicionamiento es crucial para obtener el máximo beneficio de los nuevos medicamentos con el menor riesgo y coste.
Una opción que en ocasiones se propone o valora, implícita o explícitamente, es financiarlo todo a un precio lo más reducido posible y sin condiciones de uso, y que sean las propias decisiones individuales de los prescriptores las que acaben posicionando los nuevos productos en terapéutica, fiando en las leyes de un mercado que es a todas luces imperfecto. Es lo que ocurre en sistemas sanitarios que carecen, de forma voluntaria o por una gestión deficiente, de un posicionamiento posregulatorio efectivo, ya sea a nivel estatal, regional o local (comisiones de farmacia). Aparte de las carencias para guiar la introducción en terapéutica que ya hemos identificado en la información regulatoria de los nuevos medicamentos, los mayores problemas de este modelo naïf es que traslada la variabilidad a nivel individual y deja el campo totalmente abierto a la influencia de la promoción comercial. El posicionamiento, decidiendo el lugar en terapéutica, es algo que se realizará siempre, bien a nivel estatal, regional, local o individual. Los problemas de variabilidad que a menudo se exponen cuando las decisiones se toman a nivel local o regional8, no disminuirán precisamente prescindiendo de órganos de decisión multidisciplinares y dejando el posicionamiento de los nuevos fármacos a nivel individual. Es más, la variabilidad de decisiones terapéuticas en escenarios de elevada incertidumbre, relativamente comunes para los nuevos tratamientos, podría aumentar si toda decisión recae en el nivel individual del prescriptor. Por supuesto, el posicionamiento centralizado también ha de dejar al prescriptor la adaptación terapéutica correspondiente al nivel individual del paciente.
Por otra parte, el «posicionamiento» y el «nicho» de mercado son conceptos paralelos en el marketing farmacéutico. Como es bien conocido, y sucede desde hace tiempo9, la formación y actualización en los profesionales de la salud está en gran parte financiada10 y, por tanto, mediatizada (selección de programas, ponentes e introducción de ideas sesgadas) por la industria farmacéutica. Aparte de la promoción directa, se costean congresos, sesiones de formación, cursos y jornadas. Incluso un 75% de la formación desarrollada por las propias sociedades científicas tiene este origen en su financiación11. También se costea información con tintes promocionales difícilmente identificables en medios de comunicación sanitarios y generalistas12. La mayoría de los autores de guías de práctica clínica presentan conflictos de intereses económicos con la industria sobre cuyos fármacos están aportando recomendaciones13. Se financia a asociaciones de pacientes14 y a los propios centros sanitarios para equipamiento, investigación, formación, etc. Lo que las compañías invierten en promoción supera con creces a sus inversiones en I + D15, y recae asimismo en el precio. España no está precisamente a la cola de la inversión farmacéutica en promoción: la inversión promocional en España supera a la del Reino Unido16, y los pagos de la industria a médicos (181 millones €) superan a la suma de Reino Unido (58 millones) y Alemania (109 millones)17. Obviamente, tan enorme esfuerzo sería absurdo si no influyese en la prescripción y, en consecuencia, en el nicho que el fármaco acaba ocupando en terapéutica. Por ejemplo, se ha comprobado que los conflictos de intereses en documentos de recomendación (desde guías de práctica clínica a consensos de expertos) se relacionan con posicionamientos favorables a la terapia del financiador18.
En esta situación, no exenta de críticas por la sociedad civil19, confiar en que este mercado (imperfecto) se autorregule sin una gestión bien informada de los sistemas sanitarios, puede resultar una quimera muy onerosa, capaz de dañar gravemente la sostenibilidad del propio sistema sanitario público, además de poder perjudicar a determinados pacientes, utilizando novedades que no hayan demostrado aportar realmente beneficio en salud frente al estándar y sí los expongan a toxicidad. Si se quiere gestionar una adecuada introducción de las novedades terapéuticas en el mercado, es imprescindible considerar su posicionamiento terapéutico. Esto se basa necesariamente en una evaluación posregulatoria complementaria, pero va más allá. No basta con reunir, aportar y analizar más resultados y comparaciones, hay que aplicarlos prudentemente a la práctica clínica actual y especificar un lugar en terapéutica ideal para el nuevo fármaco, con el fin de obtener los mejores resultados en salud para los pacientes.
Establecer el escenario de utilidadEl sistema público de salud necesita plantearse específicamente, en el marco de la indicación aprobada, para qué pacientes concretos interesa utilizar el medicamento, en qué situaciones y con qué alternativas competiría, bien como preferente o como similar, antes de considerar criterios económicos. Por eso se puede decir que se trata de un «preposicionamiento» previo e independiente de la valoración económica, pues el posicionamiento final ha de realizarse tras la evaluación económica y fijación de precios. Más adelante se aportan algunos ejemplos que ayudan a comprender mejor esta cuestión del escenario de utilidad/preposicionamiento y su importante alcance real.
El lugar en terapéutica, asignado al nuevo tratamiento frente al estándar, pivota sobre el hecho de que presente o no un beneficio clínico adicional relevante20. Varios países y organizaciones de nuestro entorno emplean clasificaciones del beneficio clínico para la evaluación terapéutica posregulatoria comparativa (tabla 2).
Clasificaciones del beneficio clínico para la evaluación terapéutica posregulatoria comparativa en países y organizaciones de nuestro entorno
| Organismo | Clasificación |
|---|---|
| AIFA, Agencia Italiana de Medicamentos21 | Valor terapéutico añadido:1. máximo2. importante3. moderado4. escaso5. ausente |
| IQWIG, Alemania22 | Magnitud del beneficio adicional:1. mayor (mejora grande y sostenida)2. considerable (mejora notable)3. menor (mejora moderada pero no marginal)[1–3 implican beneficio clínico adicional relevante]4. no cuantificable5. beneficio adicional no probado6. beneficio inferior (menor que el comparador) |
| HAS, Francia23,24 | Mejora del servicio médico prestado (ASMR, siglas en francés):I. mayorII. importanteIII. moderadoIV. menorV. inexistente; significa «ausencia de progreso terapéutico» |
| ESMO-MCBS (oncología)25 | Magnitud del beneficio clínico:Escenarios curativos: A-B-CEscenarios no curativos: 1–2–3-4-5A, B y 4, 5 se consideran beneficios sustanciales |
AIFA: Agenzia Italiana del Farmaco. IQWIG: Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen. HAS: Haute Autorité de Santé. ESMO-MCBS: European Society of Clinical Oncology - Magnitude of Clinical Benefit Scale.
La figura 1A ilustra a grandes rasgos el modelo propuesto de evaluación y posicionamiento. Este modelo se empezó a seguir en España a partir de la introducción de REvalMed en 202126, aunque actualmente se ha detenido. Con la información del beneficio/riesgo comparativo frente al estándar, se establece el escenario de utilidad. En la figura 1B se esquematiza el resultado de aplicar esta evaluación terapéutica posregulatoria a un nuevo tratamiento. Dentro de la indicación autorizada pueden existir situaciones o subpoblaciones con beneficio clínico mayor, menor o nulo frente a las alternativas terapéuticas ya existentes, así como escenarios de incertidumbre. Frente a los comparadores, que pueden ser varios y distintos en diferentes subpoblaciones, la nueva terapia puede presentar un beneficio clínico adicional relevante y considerarse preferente a ellos, o no. En caso de que no haya diferencias relevantes en el beneficio, los tratamientos considerados pueden considerarse alternativas de similar beneficio clínico (de forma que se pueda elegir indistintamente entre ellas), o bien tratarse simplemente de opciones terapéuticas, cuando sus importantes diferencias no permiten elegir cualquiera de ellas, sino que se precisa una selección más cuidadosa y adaptada a cada paciente, ya sea por diferente perfil de seguridad, forma de administración, comodidad, etc. (fig. 1C).
 Secuencia de procedimientos para la evaluación y posicionamiento de nuevos medicamentos/indicaciones. B) Escenario de utilidad. C) Opciones para la clasificación relativa respecto a las opciones disponibles.")
Preposicionamiento, previo a la evaluación económica y análisis de impacto presupuestario, como resultado de la aplicación de una evaluación posregulatoria comparativa.
A) Secuencia de procedimientos para la evaluación y posicionamiento de nuevos medicamentos/indicaciones. B) Escenario de utilidad. C) Opciones para la clasificación relativa respecto a las opciones disponibles.
Esto se complica, con importante incertidumbre añadida, porque a menudo (en el 34% de los casos), el nuevo medicamento no se ha comparado con el estándar actual27, lo que obliga a comparaciones indirectas ajustadas. También es posible que la magnitud del beneficio sea diferente en subpoblaciones o subgrupos28.
La cuarta garantía (posicionamiento terapéutico y eficiencia)Todo medicamento autorizado debe haber sido capaz de superar lo que se denominaron en principio, desde la perspectiva comercial, las «3 barreras» (three hurdles), que consisten en demostrar calidad, seguridad y eficacia29. Desde la perspectiva sanitaria, es decir, del paciente a quien se orientan, deberíamos llamarlas más propiamente «garantías» básicas. Los sistemas de salud más avanzados y con cobertura pública han añadido la «cuarta garantía»30,31, con la que se pretende asegurar que los medicamentos sean además coste-efectivos29. Esto confluye con la definición de la OMS del uso racional del medicamento, que incluye la cuestión del coste32, imprescindible para el acceso de los pacientes a los tratamientos33. Si bien la cuarta garantía no forma parte del ámbito regulatorio, debe ser abordada igualmente de forma obligatoria, como parte del proceso de decisión acerca de la financiación de medicamentos (Ley de Garantías)34.
Una adecuada evaluación económica y de impacto presupuestario no puede hacerse de forma simplista, valorando la comparación aportada por el ensayo pivotal en toda la indicación autorizada. Como ya hemos visto, puede haber otros comparadores, a menudo no hay una comparación directa con el estándar y es frecuente que la indicación autorizada no sea suficientemente específica para definir con precisión la población diana.
Todo esto requiere tomar decisiones sobre el escenario de utilidad y evaluar correctamente los inputs para diseñar adecuadamente la evaluación económica. La complejidad de estas decisiones implica una evaluación de la evidencia, una interpretación clínica certera y una aplicación razonable y consensuada, adecuada a la realidad de la terapéutica actual en el sistema sanitario. Aunque en los estudios económicos esto puede ser abordado en las fases iniciales por los propios autores, para una evaluación económica que pretenda sustentar la financiación pública en todo un país, el escenario de utilidad debería ser definido por un consenso multidisciplinar de expertos (evaluadores y clínicos)35, expresando las expectativas concretas del sistema sanitario sobre el uso que se le quiere dar a la nueva terapia. Esto es aún más importante teniendo en cuenta que la evaluación económica puede ser encargada al propio laboratorio, con intereses propios sobre el nicho terapéutico que ocupe su fármaco20. Por tanto, el análisis económico para el sistema sanitario público, bien sea realizado por evaluadores independientes, o bien sea encargado al laboratorio y posteriormente revisado por evaluadores independientes, ha de realizarse partiendo del consenso sobre el escenario de utilidad y las alternativas o comparadores.
Finalmente, el financiador deberá tener en cuenta tanto el lugar en terapéutica ideal como sus incertidumbres y la cuestión económica, con el fin de negociar el precio y decidir si se puede (y si se debe) financiar. En caso positivo, habrá también que considerar si se hace en todo el escenario de utilidad preespecificado (fig. 1A), o es preciso restringirlo por cuestiones de eficiencia o de impacto presupuestario (fig. 2). Un acuerdo de precios en todo el escenario de utilidad supone la situación potencialmente más favorable para el financiador y para el laboratorio, con un precio razonable extensible a todos los pacientes que se pueden beneficiar clínicamente. En cambio, al financiarse con restricciones sobre el escenario de utilidad, el financiador no se beneficia de todo el potencial terapéutico del medicamento, ya que pacientes que podrían beneficiarse, quedan sin tratar. Por su parte, el laboratorio probablemente tampoco obtiene todas sus ganancias potenciales. Aunque el laboratorio también pierda, esta situación se da a veces porque carece de flexibilidad negociadora, debido a la rigidez de posiciones centrales de la empresa, que pivotan sobre precios más altos conseguidos en otros países. En sistemas sanitarios públicos con amplia financiación y cobertura universal, en los cuales los laboratorios tienen dificultades para adaptar el precio es importante, a menos que la financiación esté delimitada por el escenario de utilidad.
, un coste utilidad que no supere el umbral supone financiación; si supera el umbral supone renegociación del precio y restricciones adicionales según Patient Access Scheme (condiciones de acceso para pacientes).")
Modelo de financiación selectiva en función del precio e impacto presupuestario para nuevos medicamentos/indicaciones con beneficio clínico adicional.
*Situación difícil de asumir para los sistemas sanitarios cuando el nuevo fármaco presenta beneficio clínico adicional relevante. **En el entorno NICE (National Institute for Health and Care Excellence; Inglaterra y Gales), un coste utilidad que no supere el umbral supone financiación; si supera el umbral supone renegociación del precio y restricciones adicionales según Patient Access Scheme (condiciones de acceso para pacientes).
Para ilustrar lo indicado, seguidamente se presentan ejemplos actuales de alto impacto sanitario y económico. En ellos se observa que establecer el escenario de utilidad en una evaluación posregulatoria, resulta imprescindible para lograr un uso adecuado de los fármacos, de forma que los pacientes obtengan el máximo beneficio clínico, y también se eviten gastos innecesarios y superfluos.
Abemaciclib en adyuvanciaAbemaciclib en adyuvancia es un caso reciente, paradigmático y de muy alto impacto clínico y presupuestario, que nos puede servir para comprobar la perspectiva de los sistemas de evaluación posregulatoria. Fue autorizado por la CE en 2022 para el tratamiento adyuvante de cáncer de mama hormonosensible, añadido a la hormonoterapia estándar. El ensayo pivotal empezó reclutando pacientes de alto riesgo según una valoración clínica de la enfermedad (cohorte 1; 90% de las pacientes). Tras una modificación en el protocolo, incluyó también un 10% de pacientes con un valor alto del índice de riesgo Ki-67 ≥ 20% (cohorte 2). El propio Comité de medicamentos de uso humano (CHMP, por sus siglas en inglés) de la EMA, consideró que solo estaba demostrado el beneficio en la cohorte 1, según consta en el European Public Assessment Report35, es decir, en pacientes que presentaban 4 o más ganglios afectados, o 1–3 y al menos uno de los siguientes criterios: tumor ≥5 cm o grado histológico 3.
Sin embargo, la indicación autorizada por la CE en la ficha técnica solamente refiere pacientes que presentan «nodos positivos con alto riesgo de recurrencia», con lo cual el criterio para considerar «alto riesgo» quedaría abierto en la práctica clínica.
Por tanto, la selección de pacientes queda escasamente definida en la indicación autorizada. Nada sugiere en la ficha técnica que tal indefinición haya sido fruto de una extrapolación intencional a otros grupos de pacientes. Por el contrario, cuando se presentan los resultados de eficacia, se indican exclusivamente los de la cohorte 1, aunque no se explicita el motivo. Simplemente, lo que figura en la ficha técnica es la indicación que fue solicitada por el laboratorio, ya que el balance beneficio/riesgo en esa indicación se considera positivo36. Esto muestra la limitación en la perspectiva y misión de la agencia reguladora, que no ha considerado modificar la indicación solicitada para facilitar la identificación de los pacientes en los que se ha establecido el beneficio clínico.
Sin embargo, en la práctica clínica, es necesario identificar a los pacientes que se benefician de este fármaco. Tratar a pacientes que no se benefician implica exponerles a reacciones adversas innecesarias y agotar tempranamente el uso de abemaciclib. Este medicamento y otro de su mismo mecanismo de acción constituyen el tratamiento preferente que mejorará la supervivencia en los pacientes que recaigan más adelante37.
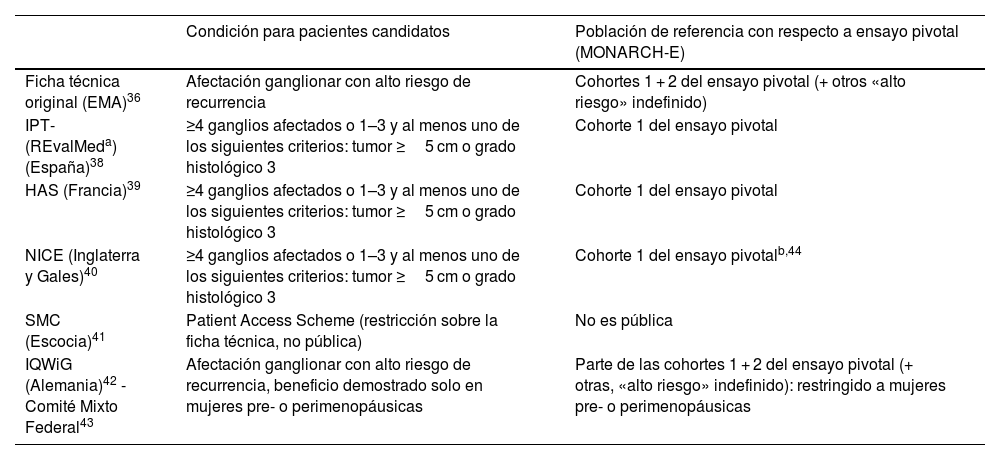
En cuanto a la evaluación económica, el coste utilidad estimado en la cohorte 1, no sería totalmente extrapolable a la población indefinida de pacientes de alto riesgo expresada en la indicación. Por su parte, el análisis de impacto presupuestario estaría infraestimado si se calculara con los criterios especificados de la cohorte 1 y luego, en la práctica, se usara abiertamente para cualquier «paciente de alto riesgo» como expresa, sin más, la indicación. En la tabla 3 se exponen las recomendaciones de los distintos organismos implicados.
Condiciones de uso de abemaciclib en adyuvancia según recomendaciones de diversos organismos, dentro de la indicación para pacientes con cáncer de mama con receptores hormonales positivos y HER2
| Condición para pacientes candidatos | Población de referencia con respecto a ensayo pivotal (MONARCH-E) | |
|---|---|---|
| Ficha técnica original (EMA)36 | Afectación ganglionar con alto riesgo de recurrencia | Cohortes 1 + 2 del ensayo pivotal (+ otros «alto riesgo» indefinido) |
| IPT-(REvalMeda) (España)38 | ≥4 ganglios afectados o 1–3 y al menos uno de los siguientes criterios: tumor ≥5 cm o grado histológico 3 | Cohorte 1 del ensayo pivotal |
| HAS (Francia)39 | ≥4 ganglios afectados o 1–3 y al menos uno de los siguientes criterios: tumor ≥5 cm o grado histológico 3 | Cohorte 1 del ensayo pivotal |
| NICE (Inglaterra y Gales)40 | ≥4 ganglios afectados o 1–3 y al menos uno de los siguientes criterios: tumor ≥5 cm o grado histológico 3 | Cohorte 1 del ensayo pivotalb,44 |
| SMC (Escocia)41 | Patient Access Scheme (restricción sobre la ficha técnica, no pública) | No es pública |
| IQWiG (Alemania)42 -Comité Mixto Federal43 | Afectación ganglionar con alto riesgo de recurrencia, beneficio demostrado solo en mujeres pre- o perimenopáusicas | Parte de las cohortes 1 + 2 del ensayo pivotal (+ otras, «alto riesgo» indefinido): restringido a mujeres pre- o perimenopáusicas |
EMA: European Medicines Agency; IPT: Informes de Posicionamiento Terapéutico; HAS: Haute Autorité de Santé; NICE: National Institute for Health and Care Excellence; SMC: Scottish Medicines Consortium; IQWiG: Institut für Qualität und Wirtschaftlichkeit im Gesundheitswesen.
Procedimiento de evaluación y posicionamiento con la incorporación de nodos multidisciplinares, vigente en España entre 2021 y julio de 2023.
El modelo económico del laboratorio presentado al NICE incluyó las cohortes 1 y 2, pero tras la revisión se exigió restringir el modelo a la cohorte 1, por considerarse más representativa de lo esperable en el Reino Unido44.
En consecuencia, realizar una evaluación posregulatoria que especifique el verdadero escenario de utilidad de abemaciclib en adyuvancia es importante para guiar su incorporación en la terapéutica, así como para definir el escenario y los «inputs» de la evaluación económica y el análisis de impacto presupuestario. El impacto asistencial y económico de esto es elevado: el cáncer de mama es uno de los de mayor incidencia anual (el primero en mujeres), el subtipo hormonal es el más prevalente, el escenario es precoz, donde el número de pacientes es mayor que en estadios avanzados, y el precio del tratamiento con abemaciclib sobrepasa los 1.500 € mensuales.
Pembrolizumab en adyuvancia en cáncer de pulmónSe ha publicado un ensayo clínico comparativo de pembrolizumab, un tratamiento inmunoterápico para el cáncer de pulmón en neoadyuvancia más adyuvancia. Muestra resultados claramente beneficiosos frente a la quimioterapia sola, al reducir la proporción de pacientes que recaen o progresan tras la cirugía. Ha sido autorizado por la FDA y por la CE.
Sin embargo, meses antes se autorizó en Europa nivolumab en neoadyuvancia, que se comparó en un ensayo frente a quimioterapia sola. Pembrolizumab y nivolumab no se han comparado entre sí, pero los resultados hacen esperar que pembrolizumab no sería superior a nivolumab, si bien este último solo es eficaz en pacientes con el marcador PD-L1 positivo (≥1%)45. Una evaluación posregulatoria de pembrolizumab, dividiría esta indicación en 2 partes: en pacientes con PD-L1 negativo (<1%), se compararía con quimioterapia, donde saldría como preferente, en virtud del estudio pivotal; en pacientes con PD-L1 ≥ 1%, la eficacia sería similar a nivolumab en neoadyuvancia, pero al no tener necesidad de administrarse tanto tiempo como pembrolizumab, nivolumab sería más seguro y no afectaría tanto a la posibilidad de que el tumor siga siendo sensible a la inmunoterapia si hay recaída. Por tanto, podría ser preferente en el balance beneficio/riesgo. Existe incertidumbre sobre el beneficio de la adyuvancia si se ha administrado neoadyuvancia previa, especialmente en pacientes con respuesta patológica completa46.
Las consecuencias de este preposicionamiento para los pacientes, pendientes de una comparación indirecta ajustada confirmatoria, podrían ser una mayor seguridad, y es razonable esperar una mayor efectividad del tratamiento en los que recaigan, al poder reintroducir con más expectativas la inmunoterapia. Para el análisis económico, este preposicionamiento definiría 2 escenarios, según el PD-L1. El impacto presupuestario de pembrolizumab en pacientes con PD-L1 < 1%, al usarse durante más tiempo (neoadyuvancia más adyuvancia), podría alcanzar 120 millones de euros. El de nivolumab sería de 23 millones45.
Los casos de abemaciclib y pembrolizumab, así como los ejemplos expuestos en la tabla 1, son solo algunos, entre muchos, casos en los que se necesita una concreción del escenario de utilidad más allá del texto de la indicación. Así, por ejemplo, es conocido el caso de atalureno en distrofia muscular de Duchenne: su ensayo no mostró beneficio y el CHMP no avaló su autorización, pero la CE concedió una aprobación condicionada a un segundo estudio en un subgrupo de pacientes. Este segundo estudio se hizo y de nuevo falló. El Informe de Posicionamiento Terapéutico (IPT) dejó claros estos fallos para demostrar utilidad47, lo cual fue probablemente determinante para no financiarlo en España; el coste era superior a 150.000 € anuales. Ha sido retirado 8 años después en Europa48, pero las ventas mundiales del laboratorio de este medicamento en 2022 ascendieron a 288,6 millones de dólares49.
Tremelimumab/durvalumab en hepatocarcinoma ha mostrado ser mejor que sorafenib, un tratamiento que ya había quedado obsoleto, aunque esta es la comparación que se presenta en la ficha técnica. Una comparación indirecta ajustada sugiere que no sería mejor que atezolizumab+bevacizumab, el estándar50 actual. La decisión se complica al recibir durvalumab en monoterapia una opinión positiva del CHMP para esta indicación51, basándose en una tercera rama del mismo ensayo pivotal.
En insuficiencia cardiaca, sacubitril/valsartan52 y dapagliflozina53 presentan concreciones clave en sus IPT que ayudan a introducirlos adecuadamente en aquellos pacientes que los necesitan porque han fallado a otros tratamientos, lo cual no se especifica en la ficha técnica.
Para cáncer de endometrio en pacientes con biomarcadores para inestabilidad de microsatélites o deficiencias en la reparación del ADN, la indicación autorizada en Europa permitiría usar pembrolizumab en monoterapia o combinado con lenvatinib. Sin embargo, carecemos de una comparación entre ambas opciones54. Si no se pone de manifiesto una ventaja en eficacia por añadir lenvatinib, pembrolizumab en monoterapia supondría menos toxicidad para las pacientes y menor coste para el sistema.
ConclusiónLa forma en que se incorporan los nuevos medicamentos en la terapéutica es un punto clave para la sostenibilidad de los sistemas sanitarios públicos europeos. Esto implica evaluarlos para posicionarlos, es decir, establecer su lugar en terapéutica frente a las demás opciones.
Un posicionamiento adecuado consta de 2 fases. En una primera fase, independiente del coste, se establece un preposicionamiento con el escenario de utilidad del fármaco. Además, se ha de considerar si el fármaco pasaría a ser preferente, constituiría una alternativa terapéutica de similar beneficio clínico, o sería una opción adicional en situaciones específicas.
La segunda fase es la que se inicia con la evaluación económica, partiendo del escenario de utilidad clínica. Con la evaluación económica, la mayor aproximación posible a los costes de fabricación y desarrollo del fármaco y el análisis de impacto presupuestario, se procede a la negociación de precio y financiación, tras la cual se realizará el posicionamiento final.
Idealmente, el posicionamiento final, tras la negociación del precio, sería idéntico al escenario de utilidad considerado en la fase de evaluación clínica, pero precios muy elevados pueden conllevar una negativa de financiación o una cobertura restringida exclusivamente para los pacientes que obtienen un beneficio más relevante o que presentan más certeza en cuanto al mismo.
Por tanto, la evaluación terapéutica posregulatoria, que permite establecer el escenario de utilidad terapéutica de un nuevo fármaco, sus incertidumbres y un preposicionamiento comparativo, resulta de enorme interés, tanto para diseñar la evaluación económica posterior, como para guiar la introducción del nuevo fármaco en terapéutica. Tiene un doble componente, de evaluación para el posicionamiento: no solo se trata de mostrar la evidencia de beneficio frente al estándar y sus incertidumbres, como se prevé con la evaluación centralizada europea, sino que, partiendo de ella, valorándola y aplicándola con el mejor criterio clínico y evaluador, implica tomar decisiones que ubiquen lo mejor posible a los nuevos tratamientos en la práctica clínica actual. Tras el desafortunado desmantelamiento de REvalMed, es imprescindible que la política farmacéutica española recupere esta dimensión que se ha conocido como «la cuarta garantía», si se quiere gestionar la introducción de las nuevas terapias en la sanidad pública de forma razonable y eficiente, guiada por el beneficio clínico para los pacientes.
FinanciaciónLos autores declaran que no hubo financiación para la elaboración de este artículo.
Conflicto de interesesManuel Jesús Cárdenas Aranzana ha participado en «advisory boards» para Merck Serono e Incyte.
AutoríaEmilio Alegre concibió el trabajo y redactó un texto previo (preborrador) que circuló en septiembre 2023 a Silvia Fénix, Dolores Fraga, Francesc Puigventós y Eduardo López Briz, que valoraron la oportunidad e interés del trabajo. En diciembre y enero se reenvió a todos el primer borrador y se añadió a Manuel Cárdenas y Carmen María Domínguez. Todos ellos lo revisaron e hicieron comentarios y aportaciones relevantes, incluyendo modificaciones, supresiones y adicciones al texto y a las tablas/figuras, así como a la bibliografía de referencia. Todos los autores han revisado y aprobado la versión final.
Declaración de contribución de autoría CRediTEmilio Jesús Alegre-del Rey: Writing – review & editing, Writing – original draft, Visualization, Validation, Supervision, Project administration, Methodology, Formal analysis, Conceptualization. Silvia Fénix-Caballero: Writing – review & editing, Validation, Methodology, Formal analysis, Conceptualization. María Dolores Fraga Fuentes: Writing – review & editing, Visualization, Validation, Methodology, Formal analysis. Manuel Jesús Cárdenas Aranzana: Writing – review & editing, Visualization, Methodology, Formal analysis. Eduardo Lopez-Briz: Writing – review & editing, Visualization, Validation, Methodology, Formal analysis, Conceptualization. Francesc Puigventós Latorre: Writing – review & editing, Visualization, Validation, Supervision, Formal analysis, Conceptualization. Carmen María Domínguez-Santana: Writing – review & editing, Visualization, Validation, Methodology, Formal analysis.

