




El síndrome de realimentación (SR) es un cuadro clínico complejo que ocurre como consecuencia de la reintroducción de la nutrición (oral, enteral o parenteral) en pacientes malnutridos. Los pacientes presentan trastornos en el balance de fluidos, anomalías electrolíticas –como hipofosfatemia, hipopotasemia e hipomagnesemia– alteraciones en el metabolismo hidrocarbonado y déficits vitamínicos. Esto se traduce en la aparición de complicaciones neurológicas, respiratorias, cardíacas, neuromusculares y hematológicas. En este artículo se han revisado la patogenia y las características clínicas del SR, haciendo alguna sugerencia para su prevención y tratamiento. Lo más importante en la prevención del SR es identificar a los pacientes en riesgo, instaurar el soporte nutricional de forma prudente y realizar una corrección adecuada de los déficits de electrolitos y vitaminas.
Refeeding syndrome is a complex syndrome that occurs as a result of reintroducing nutrition (oral, enteral or parenteral) to patients who are starved or malnourished. Patients can develop fluid-balance abnormalities, electrolyte disorders (hypophosphataemia, hypokalaemia and hypomagnesaemia), abnormal glucose metabolism and certain vitamin deficiencies. Refeeding syndrome encompasses abnormalities affecting multiple organ systems, including neurological, pulmonary, cardiac, neuromuscular and haematological functions. Pathogenic mechanisms involved in the refeeding syndrome and clinical manifestations have been reviewed. We provide suggestions for the prevention and treatment of refeeding syndrome. The most important steps are to identify patients at risk, reintroduce nutrition cautiously and correct electrolyte and vitamin deficiencies properly.
Usamos el término síndrome de realimentación (SR) para describir las alteraciones metabólicas que ocurren durante la repleción nutricional, ya sea oral, enteral o parenteral, de individuos severamente desnutridos o privados de alimento1. El hecho fundamental del SR es la hipofosfatemia severa2, que se acompaña de anomalías en el balance de fluidos, alteraciones en el metabolismo hidrocarbonado y ciertos déficits vitamínicos, por ejemplo de tiamina, así como de hipopotasemia e hipomagnesemia2. Esto se traduce clínicamente en la aparición de, entre otras, alteraciones neurológicas, respiratorias, cardiovasculares y hematológicas pocos días después del inicio de la realimentación, lo que conlleva un aumento de la morbilidad e incluso mortalidad del paciente3.
El estudio clásico que describe el SR fue realizado por Keys et al4 en varones sanos objetores de conciencia de la Segunda Guerra Mundial. Los participantes se sometían a semiayuno durante 6 meses, tras los que se reintroducía una alimentación oral normal. La consecuencia fue una disminución de la reserva cardiovascular, con insuficiencia cardíaca en algunos casos. Resultados clínicos similares pudieron verse al reinstaurar una ingesta normal en individuos que habían sufrido asedio o habían estado internados en campos de concentración en la Segunda Guerra Mundial5. Posteriormente, con la aparición de la nutrición parenteral (NP) y enteral (NE) se observó el mismo tipo de complicaciones en pacientes desnutridos que recibieron soporte nutricional agresivo6.
Además del SR, sobre todo en el paciente ingresado y grave, existen numerosas causas de hipofosfatemia, hipomagnesemia e hipopotasemia. Las más comunes se enumeran en la tabla 1 y deberán tenerse en mente al hacer el diagnóstico diferencial.
Causas de hipofosfatemia, hipomagnesemia e hipopotasemia
|
El SR es un fenómeno común en pacientes desnutridos con depleción previa de masa corporal magra. Su incidencia varía según las series y los criterios diagnósticos utilizados. Hernández Aranda et al7 encontraron una incidencia de SR del 48 % en una cohorte de 148 pacientes con desnutrición de leve a severa a los que se les administró soporte nutricional. González et al8 diagnosticaron SR al 25 % de 107 pacientes oncológicos que recibieron NP o NE; la incidencia fue más alta en el subgrupo que recibió NE. Flesher et al9 valoraron la aparición de alteraciones propias del SR en 51 pacientes con NE en los que se alcanzó el objetivo nutricional en un tiempo de tan sólo 17 h. El 80 % de los pacientes presentaron depleción de fosfato, magnesio o potasio después del inicio de la NE. La incidencia osciló entre el 74 %, en el grupo de pacientes "no en riesgo", y el 93 % en el grupo "en riesgo"9 (tabla 2).
Pacientes en riesgo de desarrollar síndrome de realimentación
|
EPOC: enfermedad pulmonar obstructiva crónica.
La hipofosfatemia es un hallazgo relativamente frecuente en los pacientes hospitalizados; afecta al 3-42 % de ellos. La incidencia es especialmente alta en las unidades de cuidados intensivos y de enfermedades infecciosas10. Marik y Bedigian11 detectaron hipofosfatemia relacionada con la realimentación en el 34 % de los pacientes de cuidados intensivos después de un ayuno de tan sólo 48 h.
FisiopatologíaLos tejidos utilizan de forma preferente los hidratos de carbono para obtener energía. Nuestro organismo dispone de una reserva limitada, en forma de glucógeno almacenado en el hígado y el músculo. Durante el período inicial de ayuno se utilizan como fuente energética los depósitos de glucógeno. Tras el agotamiento de estos depósitos se inicia la proteólisis, que suministra aminoácidos para la gluconeogénesis, lo que permitirá disponer de glucosa a los tejidos dependientes de la misma (cerebro, médula renal, células rojas). Después de 72 h de ayuno y con el objetivo de evitar la movilización de proteínas del músculo esquelético, las rutas metabólicas derivan hacia la lipólisis, con obtención de ácidos grasos libres. Estos ácidos grasos libres pueden seguir dos rutas: a nivel periférico son utilizados por las células para producir energía, y a nivel hepático se usan como sustratos para la síntesis de cuerpos cetónicos (ácido acetoacético, ácido β-hidroxibutírico y acetona) mediante la cetogénesis. El cerebro puede utilizar los cuerpos cetónicos, que atraviesan la barrera hematoencefálica, como fuente energética. Sin embargo, la capacidad de las células para oxidar cuerpos cetónicos es limitada, lo que conduce a una situación de cetosis y finalmente acidosis metabólica. Durante este proceso adaptativo, junto a la respuesta metabólica descrita, ocurre también una serie de cambios hormonales encaminados a mantener las funciones vitales: descenso de insulina y aumento de glucagón12, aumento de la secreción de hormona de crecimiento (acción lipolítica y cetogénica), descenso del factor de crecimiento similar a la insulina (IGF-1), descenso de triyodotironina, aumento de la secreción de cortisol, descenso de las concentraciones de leptina13,14 y aumento de las catecolaminas. Todo este cambio hormonal conduce a un descenso del metabolismo basal y aumenta la disponibilidad de ciertos sustratos energéticos.
En el ayuno, además de la pérdida de peso se produce una disminución de la masa celular y un aumento del agua extracelular. Los valores plasmáticos de electrolitos como el potasio, el fósforo y el magnesio se mantienen dentro de la normalidad; sin embargo, su contenido corporal total está disminuido.
La patogénesis del SR es compleja dado que intervienen cambios metabólicos y fisiológicos que se suceden durante la fase de depleción y repleción de sustratos, con las consecuentes desviaciones compartimentales de electrolitos, alteraciones en el metabolismo de la glucosa y vitaminas y en el manejo del agua corporal. Cuando se reintroduce la alimentación, sobre todo si se basa en hidratos de carbono, se produce un aumento en la secreción de insulina que favorece el anabolismo y la entrada de ciertos elementos (fósforo, potasio y magnesio) al interior celular, originando un descenso de sus concentraciones plasmáticas15. El mecanismo exacto por el que ocurre el desequilibrio de fluidos es desconocido, pero se cree que la retención de agua y sodio puede deberse a un efecto antinatriurético de la hiperinsulinemia16. La tiamina es un cofactor esencial en el metabolismo de los carbohidratos17, por tanto, el aporte de una cantidad elevada de éstos incrementa su demanda. Aunque es difícil establecer si la deficiencia de tiamina se debe a la realimentación o al estado de ayuno, los pacientes desnutridos están en riesgo de desarrollar déficit y sus complicaciones asociadas. Se produce también un aumento en la conversión de T4 a T318,19, que origina un incremento en el gasto energético. En la figura 1 se muestra la fisiopatología del SR.
Manifestaciones clínicas
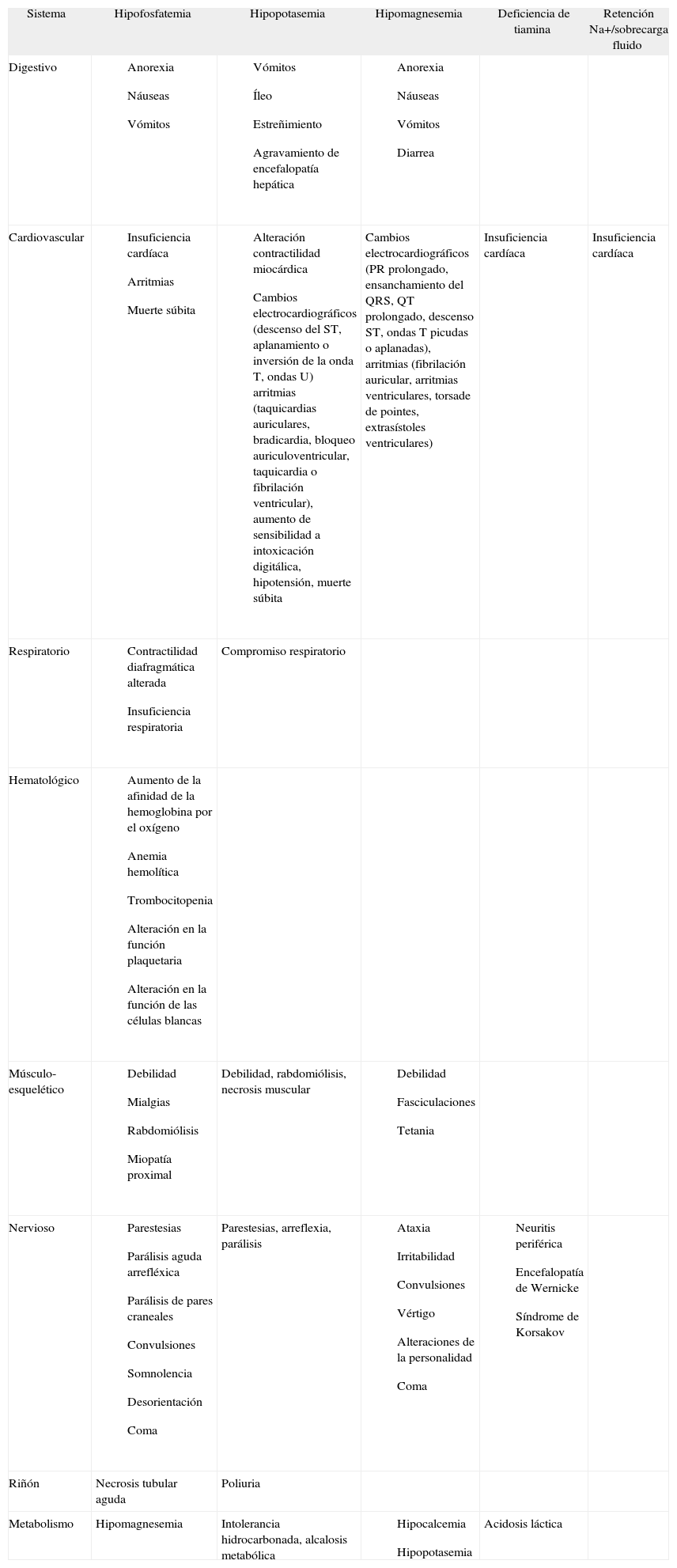
Las manifestaciones clínicas del SR se derivan de los efectos de las alteraciones hidroelectrolíticas y déficits vitamínicos descritos sobre los distintos sistemas y órganos (tabla 3).
Manifestaciones clínicas del síndrome de realimentación
| Sistema | Hipofosfatemia | Hipopotasemia | Hipomagnesemia | Deficiencia de tiamina | Retención Na+/sobrecarga fluido |
| Digestivo |
|
|
| ||
| Cardiovascular |
|
| Cambios electrocardiográficos (PR prolongado, ensanchamiento del QRS, QT prolongado, descenso ST, ondas T picudas o aplanadas), arritmias (fibrilación auricular, arritmias ventriculares, torsade de pointes, extrasístoles ventriculares) | Insuficiencia cardíaca | Insuficiencia cardíaca |
| Respiratorio |
| Compromiso respiratorio | |||
| Hematológico |
| ||||
| Músculo-esquelético |
| Debilidad, rabdomiólisis, necrosis muscular |
| ||
| Nervioso |
| Parestesias, arreflexia, parálisis |
|
| |
| Riñón | Necrosis tubular aguda | Poliuria | |||
| Metabolismo | Hipomagnesemia | Intolerancia hidrocarbonada, alcalosis metabólica |
| Acidosis láctica |
El fosfato es el principal anión intracelular. Los depósitos de fosfato en el organismo de un adulto medio son de aproximadamente 700 g, de los cuales el 80 % se localiza en el esqueleto, alrededor de un 20 % en tejidos blandos y músculo2 y sólo un 1 % en el líquido extracelular. La ingesta media de fósforo en la dieta occidental es de 1.000 a 1.400 mg diarios. Se absorbe un 80 % del fósforo ingerido, la mayoría a nivel de yeyuno por transporte pasivo, y un pequeño porcentaje mediante transporte activo dependiente de la vitamina D20. La principal vía de eliminación del fósforo es el riñón; el 90 % del fósforo se excreta por vía urinaria y sólo un 10 % por vía gastrointestinal21.
El nivel de fósforo sérico normal se mantiene en el estrecho margen que va de 2,5 a 4,5 mg/dl (1 mg/dl = 0,32 mmol/l), aunque su concentración no siempre refleja el contenido corporal total. La regulación a corto plazo de las cifras de fósforo se alcanza mediante el flujo transcelular entre los compartimentos intracelular y extracelular, dependiente de la ingesta de carbohidratos y lípidos, y de las modificaciones en el equilibrio ácido-base22. El músculo esquelético y el hueso son reservorios endógenos de fósforo; así, en caso de hipofosfatemia el fósforo muscular se moviliza para proveer a órganos vitales (cerebro, corazón, suprarrenales, riñón, tiroides, páncreas y bazo). La regulación a largo plazo depende del riñón. El fósforo se filtra en el glomérulo y el 85 % del filtrado se reabsorbe en el túbulo proximal a través del transportador de sodio-fosfato tipo 2 (NaPi-2a). La tasa de reabsorción tubular de fosfato depende del número de transportadores. La hormona paratiroidea (PTH) ocasiona la internalización y degradación intracelular de éstos, lo que supone una disminución en la reabsorción de fosfato y la aparición de fosfaturia23. Además de la PTH, también influyen otros factores hormonales en los transportadores de NaPi: insulina, glucagón, hormona de crecimiento, glucocorticoides y 1-25 dihidroxi-vitamina D24.
El fosfato es esencial para el funcionamiento celular. Tiene un papel estructural como componente de fosfolípidos, nucleoproteínas y ácidos nucleicos; desempeña un papel clave en rutas metabólicas, como la glucólisis y la fosforilación oxidativa25, y está implicado en el control de procesos enzimáticos a través de la fosforilación de proteínas. El fosfato actúa como cofactor de la gliceraldehído 3 fosfato deshidrogenasa, por lo que, en caso de hipofosfatemia, disminuye la producción de 2,3-difosfoglicerato (2,3-DPG) y de adenosina trifosfato (ATP)26. El 2,3-DPG supone el 80 % del fosfato orgánico de los eritrocitos y está implicado en la regulación de la curva de disociación del oxígeno de la hemoglobina y, por tanto, en la liberación de oxígeno a los tejidos27.
La hipofosfatemia del SR típicamente aparece en los tres primeros días tras el inicio del soporte nutricional. Los factores que se han relacionado en diversos estudios con su aparición son: hipoalbuminemia, prealbúmina < 110 g/l, y circunferencia y área muscular del brazo inferior al percentil 5. Es probable que estos factores, más que tener un papel patogénico en la aparición de la hipofosfatemia, sólo representen la gravedad de la desnutrición previa del paciente26.
La hipofosfatemia se considera severa cuando el fosfato sérico es < 126 o 1,5 mg/dl, moderada cuando los valores están en el rango de 1,5 a 2,2 mg/dl y leve si el fosfato sérico está entre 2,3 y el límite inferior de la normalidad1. Aparecen síntomas con valores de fósforo < 1,5 mg/dl, o con concentraciones mayores si el descenso es rápido, siendo muy evidentes con niveles < 1 mg/dl. La hipofosfatemia severa provoca importantes alteraciones a nivel neurológico, cardíaco, respiratorio y hematológico, y puede conducir a la muerte1. La tasa de mortalidad de los pacientes con hipofosfatemia grave es del 30 %28.
Sistema cardiovascular. Las complicaciones cardiovasculares aparecen en la primera semana de reposición nutricional. El ayuno prolongado conduce a la atrofia y la depleción de ATP de las células miocárdicas, lo que resulta en una alteración de la contractilidad. En esta situación, la reposición de líquidos junto con la retención de sodio y agua secundaria a la realimentación con hidratos de carbono, supone una sobrecarga de volumen que puede dar lugar a insuficiencia cardíaca. La hipofosfatemia severa provoca alteración de la función cardíaca en relación con la depresión de la función miocárdica debida a la depleción de ATP y por daño directo del miocardio3. O'Connor et al29 describen en pacientes con cifras de fosfato de 0,7-1,4 mg/dl un descenso en el volumen/latido, presión arterial media y gasto cardíaco, y un aumento en la presión capilar pulmonar que mejoran significativamente con la repleción de fosfato.
En este contexto, puede aparecer hipotensión, derrame pericárdico, shock, arritmias y muerte súbita. La hipofosfatemia es causa directa de arritmias ventriculares; el riesgo aumenta en caso de hipomagnesemia e hipopotasemia simultáneas30. Aparecen arritmias en hasta el 20 % de los pacientes hipofosfatémicos sin cardiopatía subyacente y en caso de infarto agudo de miocardio aumenta el riesgo de taquicardia ventricular.
Sistema hematológico. La hipofosfatemia provoca un descenso en el ATP y 2,3-DPG intraeritrocitario. El descenso del 2,3-DPG aumenta la afinidad de la hemoglobina por el oxígeno, desplazando la curva de disociación a la izquierda, con la consecuente disminución en la liberación de oxígeno a los tejidos periféricos2. La caída del ATP intraeritrocitario en un 20-50 % respecto a su valor normal provoca la aparición de esferocitosis reversible, con un incremento en la rigidez de la membrana celular31. Esto contribuye a empeorar la hipoxia tisular, ya que dificulta el paso de los eritrocitos a través de los capilares, lo que, por otra parte, condiciona la aparición de anemia hemolítica.
La hipofosfatemia severa altera la supervivencia y la función plaquetaria y puede ocasionar trombocitopenia, trastornos en la agregación plaquetaria y hemorragias secundarias. La serie blanca también se ve afectada, con trastornos de la función quimiotáctica, fagocítica y bactericida, posiblemente incrementando el riesgo de sepsis en pacientes de alto riesgo32.
Aparato respiratorio. La disfunción respiratoria en pacientes con hipofosfatemia es secundaria al descenso de la glucólisis y, probablemente, a la caída en los valores de ATP en los músculos respiratorios. La hipofosfatemia severa altera la contractilidad diafragmática33. El cuadro clínico puede presentarse como un descenso de la capacidad vital, fallo respiratorio o dificultad para desconectar a los pacientes de la ventilación mecánica.
Sistema nervioso. El mecanismo de la disfunción neurológica en la hipofosfatemia está poco claro, pero se ha sugerido que la hipoxia tisular secundaria a la anemia hemolítica y al aumento de la afinidad de la hemoglobina por el oxígeno podría ser la causa3. Las alteraciones neurológicas descritas en relación con la hipofosfatemia son: parálisis de pares craneales, parestesias, cansancio, tetania, alucinaciones, delirio, convulsiones, letargia, confusión y coma. En pacientes ocasionales puede aparecer un cuadro similar al síndrome de Guillain-Barré, que se manifiesta como una parálisis arrefléxica aguda32. En los pacientes con SR que desarrollen disfunción neurológica hay que considerar la posibilidad de una encefalopatía de Wernicke.
Sistema músculo-esquelético. La disfunción del músculo esquelético secundaria a hipofosfatemia puede manifestarse clínicamente como debilidad, mialgias, rabdomiólisis o debilidad diafragmática. Algunos pacientes presentan miopatía proximal con dificultad para la deambulación. La depleción de ATP en el miocito, y probablemente las alteraciones de la creatincinasa, provocan debilidad muscular y rotura del sarcolema con rabdomiólisis, que es especialmente común en pacientes alcohólicos con SR32. La rabdomiólisis puede conducir a la aparición de necrosis tubular aguda.
Otros. La hipofosfatemia también puede provocar manifestaciones psiquiátricas, como ansiedad o alucinaciones visuales o auditivas. A nivel gastrointestinal pueden aparecer anorexia, náuseas, vómitos o alteración de las pruebas hepáticas26. En el riñón, aumenta la excreción de magnesio, con aparición de hipomagnesemia34.
HipopotasemiaEl potasio es el principal catión intracelular. El 98 % del potasio corporal total se encuentra en el espacio intracelular. La excreción se realiza en un 80 % a través del riñón y el resto por las heces y el sudor. Tiene diversas funciones fisiológicas y participa en el mantenimiento del potencial de membrana y en la regulación de la síntesis de glucógeno y proteínas. La hipopotasemia altera el potencial de acción transmembrana, lo que resulta en una hiperpolarización de ésta con alteración de la contractilidad muscular35. El contenido de potasio corporal se regula en el riñón. La nefrona distal secreta potasio en respuesta a la aldosterona, alcalosis, dieta rica en potasio o incremento en la llegada de sodio al túbulo distal36.
Hablamos de hipopotasemia leve-moderada cuando las cifras de potasio sérico están entre 2,5 y 3,5 mEq/l. El paciente puede presentar síntomas gastrointestinales, como náuseas, vómitos, estreñimiento y también debilidad. Si no se trata puede progresar a hipopotasemia severa (potasio sérico < 2,5 mEq/l), con la aparición de disfunción neuromuscular (parálisis arrefléxica, parestesias, rabdomiólisis, necrosis muscular, compromiso respiratorio, confusión) y trastornos en la contractilidad miocárdica y la conducción de señales. La hipopotasemia severa provoca cambios electrocardiográficos, como descenso del segmento ST, aplanamiento o inversión de la onda T, o presencia de ondas U37. El paciente puede presentar arritmias cardíacas, desde taquicardia auricular, bradicardia, bloqueo auriculoventricular y extrasístoles ventriculares, hasta taquicardia y fibrilación ventricular, e incluso muerte súbita. Otras manifestaciones clínicas de la hipopotasemia son la aparición de intolerancia a la glucosa, alcalosis metabólica, empeoramiento de la encefalopatía hepática y potenciación de la toxicidad digitálica26.
HipomagnesemiaEl magnesio es el segundo catión intracelular más abundante (el más abundante de los cationes divalentes). El 99 % del magnesio corporal total se encuentra a nivel intracelular y se localiza fundamentalmente en el hueso y el músculo. Por ello, lo mismo que ocurre con el fosfato o el potasio, las determinaciones séricas no reflejan adecuadamente el magnesio corporal total o el estado del magnesio en el líquido intracelular. Sólo un 30 % del magnesio ingerido se absorbe. La absorción tiene lugar en la porción proximal del intestino delgado y es independiente de la vitamina D. El 70 % restante se elimina directamente con las heces. La excreción es fundamentalmente renal26. Actúa como cofactor de numerosas enzimas, participando en la regulación de diversas reacciones bioquímicas, como la fosforilación oxidativa.
La hipomagnesemia es frecuente en pacientes críticamente enfermos y se asocia a un incremento de la morbimortalidad. Los valores séricos normales se sitúan entre 1,8 y 2,5 mg/dl (0,65-1 mmol/l). Los pacientes con hipomagnesemia leve-moderada (magnesio sérico de 1-1,5 mg/dl) generalmente permanecen asintomáticos, no así aquellos con hipomagnesemia severa (magnesio sérico < 1 mg/dl). Las manifestaciones clínicas son diversas: disfunción neuromuscular (temblor, parestesias, tetania, hiperreflexia, fasciculaciones, convulsiones, ataxia, nistagmo, vértigo, debilidad muscular, depresión, irritabilidad, cuadros psicóticos, etc.), cambios electrocardiográficos (PR prolongado, ensanchamiento del complejo QRS, QT prolongado, descenso del ST, ondas T picudas o aplanadas), arritmias cardíacas (fibrilación auricular, taquicardia ventricular, torsade de pointes) e incluso la muerte.
Por otra parte, la hipomagnesemia puede favorecer la aparición de hipocalcemia e hipopotasemia, o complicar el tratamiento de trastornos preexistentes. La hipocalcemia inducida por hipomagnesemia es el resultado de alteraciones en la secreción y acción de la PTH sobre las células diana del hueso y el riñón38. También es característica de la hipomagnesemia la resistencia a la vitamina D, relacionada con una anomalía en la 1-α-hidroxilación de la 25(OH)D en el riñón, además de con la resistencia de los tejidos a la 1,25(OH)2D3. La hipopotasemia inducida por hipomagnesemia se debe a una alteración de la actividad Na+/K+-ATPasa, con un incremento en las pérdidas renales de este electrolito.
Deficiencia de tiaminaLa tiamina o vitamina B1 es una vitamina hidrosoluble necesaria en el metabolismo de los glúcidos, pues actúa como cofactor de la piruvato deshidrogenasa y de las transcetolasas. El déficit de tiamina provoca un aumento de la concentración de piruvato en sangre, que se transforma en lactato. Esta formación excesiva de lactato lleva a la aparición de acidosis láctica. La ingesta mínima recomendada en adultos es de 1 mg diario. La eficacia de absorción de la vitamina, mediante un mecanismo de transporte activo específico en la porción proximal del intestino delgado, es superior al 80 %. Los depósitos corporales son de unos 30 mg y se deplecionan rápidamente en caso de desnutrición. Las situaciones clínicas que se asocian con mayor frecuencia al déficit de tiamina son el alcoholismo crónico, los síndromes de malabsorción y las náuseas y vómitos del embarazo. La deficiencia de tiamina puede conducir a la aparición de insuficiencia cardíaca, encefalopatía de Wernicke (trastornos oculares, confusión, ataxia y coma) y síndrome de Korsakov (pérdida de memoria a corto plazo y confabulación)2.
La ingesta de carbohidratos aumenta las necesidades de tiamina. Se ha descrito la aparición de encefalopatía de Wernicke en pacientes desnutridos con déficit de tiamina a los que se les administraba NP con un aporte alto de hidratos de carbono39.
Retención de sodio/sobrecarga de fluidoEn las fases iniciales del SR puede aparecer retención de sodio y expansión del fluido extracelular, con el consiguiente riesgo de descompensación cardíaca40. El riesgo es mayor en los pacientes con desnutrición severa, en relación con la posible atrofia e hipocontractilidad miocárdica.
PrevenciónEl SR es una entidad clínica poco conocida en el entorno médico no especializado en nutrición y probablemente infradiagnosticada1. Los factores comúnmente asociados a la aparición del SR son la presencia de malnutrición, la reposición nutricional agresiva en las fases iniciales sin suplementación adecuada de fosfato, magnesio, potasio y tiamina, y la presencia de condiciones asociadas que exacerben el déficit de micronutrientes, minerales y electrolitos41. Para prevenir su aparición y evitar la morbimortalidad asociada existen una serie de pasos clave que se deberían seguir:
- 1.
Realizar una valoración médica y nutricional completa del paciente antes de iniciar el soporte nutricional. Esto nos permitirá identificar a los pacientes en riesgo de desarrollar SR (tabla 2).
- 2.
Monitorizar analíticamente al paciente antes y durante la realimentación, incluyendo hemograma y bioquímica completa. Los valores séricos de fósforo, magnesio y potasio pueden no reflejar sus depósitos corporales totales de forma fiable; la determinación en orina puede ser útil para detectar déficits.
- 3.
Corregir el equilibrio hídrico y las anomalías electrolíticas (especialmente, hipofosfatemia, hipopotasemia e hipomagnesemia) antes de iniciar el aporte de nutrientes. En la práctica esto supone retrasar el inicio de 12 a 24 h.
- 4.
Evitar la sobrealimentación, independientemente del método usado para estimar los objetivos calóricos. El requerimiento de glucosa mínimo de un adulto de 70 kg para suprimir la gluconeogénesis, ahorrar proteínas y aportar combustible al sistema nervioso central es de 100-150 g/día. El objetivo de aporte proteico se sitúa en 1,2-1,5 g/kg/día, aunque habrá pacientes con requerimientos aumentados o disminuidos.
- 5.
Iniciar la repleción nutricional con precaución (25 % de las necesidades calculadas en el primer día) e incrementar gradualmente el aporte hasta alcanzar el objetivo en 3-5 días1. Otros autores recomiendan empezar con 20 kcal/kg/día, o una media de 1.000 kcal/día, y aumentar el aporte de forma lenta durante la primera semana hasta que el paciente esté metabólicamente estable3.
- 6.
Suplementación empírica de electrolitos antes y durante el soporte nutricional. El incremento de la carga calórica disminuye la concentración de fósforo sérico. Se necesita proporcionar un mínimo de 10-15 mmol de fosfato por cada 1.000 kcal para mantener concentraciones séricas normales en pacientes con función renal normal. Los pacientes con desnutrición severa, enfermedades críticas, traumatismos o quemaduras pueden tener una depleción del fosfato corporal total, lo mismo que de potasio y magnesio, por lo que sus requerimientos serán más altos. Después del inicio del apoyo nutricional, los electrolitos se suplementarán en función de sus concentraciones séricas y de la respuesta al tratamiento1.
- 7.
Restricción de sodio (< 1 mmol/kg/día) y líquidos para evitar la sobrecarga de volumen. La restricción de líquidos será tal que permita mantener la función renal, sustituya las pérdidas y evite la ganancia ponderal. Los pacientes no deberán ganar más de 0,5-1 kg de peso a la semana, cualquier aumento superior a 1 kg/semana será probablemente consecuencia de la retención de fluidos42.
- 8.
Suplementación de vitaminas: los preparados multivitamínicos parenterales aportan los requirimientos recomendados por la Asociación Médica Americana. Contienen 3 o 6 mg de tiamina. Las necesidades de tiamina están aumentadas en pacientes desnutridos, alcohólicos, postoperados, sometidos a ayuno o que hayan presentado vómitos importantes. Se recomienda la administración empírica de 50-250 mg de tiamina al menos 30 min antes del inicio de la alimentación2. Debería administrarse tiamina a dosis de 50-100 mg/día por vía intravenosa (iv), o 100 mg/día por vía oral (vo) durante 5-7 días en pacientes en riesgo de déficit de esta vitamina o de desarrollar SR1. También se puede administrar a los pacientes 1-5 mg/día de ácido fólico durante 5-7 días, aunque esto no parece que vaya a prevenir el SR2. La administración diaria de un complejo multivitamínico a estos pacientes parece una práctica segura, barata y que puede prevenir complicaciones.
- 9.
Monitorización clínica estricta del paciente. El objetivo es detectar de forma temprana los datos sugestivos de SR. Deben controlarse de forma rutinaria la frecuencia cardíaca y respiratoria, la presión arterial y pulsioximetría. Es importante valorar el balance hídrico, pesar de forma regular a los pacientes y buscar en la exploración física la presencia de edema o de otros signos que indiquen sobrecarga de volumen. También es importante la detección de síntomas o signos que indiquen disfunción neuromuscular y, si es posible, debe realizarse monitorización electrocardiográfica1.
Si se diagnostica a un paciente de SR debe suspenderse de forma inmediata el soporte nutricional. El tratamiento incluirá las medidas de apoyo necesarias (tratamiento de manifestaciones cardiovasculares, respiratorias, etc.) y la corrección de las anomalías electrolíticas. También debe administrarse una dosis de 100 mg de tiamina iv en caso de cambios neurológicos. La nutrición podrá reintroducirse cuando el paciente esté asintomático y estable. Se recomienda reiniciar el apoyo nutricional a un ritmo bajo (aproximadamente el 50 % del ritmo al que se había iniciado previamente), realizar una progresión lenta a lo largo de 4-5 días, suplementar electrolitos y vitaminas de forma apropiada y monitorizar estrictamente al paciente.
Tratamiento de la hipofosfatemiaEl tratamiento de la hipofosfatemia depende de la magnitud de ésta, de la presencia o no de síntomas y de la ruta de administración de que dispongamos (enteral o parenteral)1. Algunos autores consideran que no es necesario tratarla excepto que el paciente esté sintomático o el nivel de fosfato sérico sea < 0,3 mmol/l (1 mg/ml)2. En la tabla 4 pueden verse algunas de las presentaciones farmacológicas y su composición, que podemos usar para el tratamiento de la hipofosfatemia, así como de la hipopotasemia e hipomagnesemia.
Principales presentaciones de fosfato, potasio y magnesio
| Producto | Aporte electrolítico |
| Presentaciones intravenosas | |
| Fosfato monopotásico 1 M 10 ml (fórmula magistral) | 1 mEq/ml de potasio |
| 1 mmol/ml de fosfato (30,9 mg/ml) | |
| Fosfato monosódico 1 M 10 ml (fórmula magistral) | 1 mEq/ml de sodio |
| 1 mmol/ml de fosfato (30,9 mg/ml) | |
| Glicerofosfato sódico (Meinsol KP 242) 10 ml | 2 mEq/ml de sodio |
| 1 mmol/ml de fosfato | |
| Fosfato dipotásico 1 M 10 ml | 2 mEq/ml de potasio |
| 1 mmol/ml de fosfato | |
| Cloruro potásico 1 M® 10 ml | 1 mEq/ml de potasio (38,9 mg/ml); |
| 1 mEq/ml de cloruro | |
| Cloruro potásico 2 M® 10 ml | 2 mEq/ml de potasio (77,4 mg/ml); |
| 2 mEq/ml de cloruro | |
| Acetato potásico® 1 M 10 ml | 1 mEq/ml de potasio y 1 mEq/ml de acetato |
| Ap inyect. solución cloruro potásico® vial 20 ml | 2 mEq/ml de potasio (78,25 mg/ml) |
| Cloruro potásico 18,4 %® 10 ml | 2,5 mEq/ml de potasio (96,5 mg/ml); |
| 2,5 mEq/ml de cloruro | |
| Sulfato magnésico 15 %® 10 ml (medicamento extranjero) | 1,5 g de sal |
| 1,2 mEq/ml de magnesio (0,6 mmoles/ml; 14,8 mg/ml) | |
| Presentaciones orales | |
| Fosfato sódico monobásico NM® sobres de 3,56 g de sal | 26 mmoles de fosfato (800 mg) |
| Phosphate Sandoz Forte® comp. (medicamento extranjero) | 20,4 mEq de sodio; 3,1 mEq de potasio, 16,1 mmol de fosfato (500 mg) |
| Potasion 600 mg® cápsulas | 8 mEq de potasio (313 mg) |
| Potasion solución® | 1.320 mg/5 ml de sal |
| 1 mEq/ml de potasio (39 mg/ml) | |
| Boi K® comp. efervescentes | 640 mg de sal; |
| 10 mEq de potasio (390 mg) | |
| Boi K aspártico® comp. efervescentes | 1.825 mg de sal |
| 25 mEq de potasio (975 mg) | |
| Magnesioboi® comp. | 500 mg de sal |
| 3,9 mEq de magnesio (1,95 mmoles; 47,5 mg) | |
| Actimag® solución oral | 2.000 mg/5 ml de sal |
| 14,3 mEq/5 ml de magnesio (7,15 mmoles; 170 mg) | |
| Magnesium Pyre® comp. | 570,6 mg de sal |
| 5,56 mEq de magnesio (2,78 mmoles; 67,9 mg) | |
| Magnogene® comp. | 198,84 mg de sal |
| 4,25 mEq de magnesio (2,12 mmoles; 51,4 mg) | |
Los pacientes con hipofosfatemia leve1 o moderada43, asintomáticos y con tracto gastrointestinal funcionante podrían tratarse con fosfato oral, teniendo en cuenta que puede causar diarrea. Aquellos sujetos con déficit severo, sintomáticos, o en los que no se pueda usar el tracto digestivo, recibirán suplementación iv. Las dosis que se recomiendan son empíricas, puesto que los valores séricos de fosfato no se correlacionan con los almacenes corporales totales y no existe forma de predecir la respuesta a la reposición. Esto hace necesario un seguimiento clínico y analítico muy estrecho. Una posible pauta de reposición de fosfato iv sería la administración de 0,08-0,16 mmol/kg de peso cuando el fosfato sérico es de 2,3-2,7 mg/dl; 0,16-0,32 mmol/kg en pacientes con cifras de 1,5-2,2 mg/dl y 0,32-0,64 mmol/kg si el fosfato es < 1,5 mg/dl44. La dosis calculada debe administrarse en 4-6 h, sin sobrepasar una velocidad de 7 mmol de fosfato/h. Está contraindicada la administración de fosfato iv en pacientes con hipercalcemia, por el riesgo de calcificación metastásica, o con hiperpotasemia26. Los efectos secundarios son: hiperfosfatemia, hipocalcemia, tetania, hipotensión, hiperpotasemia, hipernatremia y calcificación metastásica45. Así, algunos autores recomiendan suspender su administración cuando se alcanzan valores séricos de 1-2 mg/dl46; otros, sin embargo, continúan la suplementación hasta que el paciente está asintomático o la concentración de fosfato sérico está en el rango normal.
Tratamiento de la hipopotasemiaLa suplementación de potasio puede realizarse por vo o iv. Usaremos la ruta iv para tratar a pacientes con déficit severo, sintomáticos, o en los que no puede usarse el tracto digestivo; además, hay que tener en cuenta que el potasio oral puede provocar efectos secundarios gastrointestinales (cólicos, diarrea). Se recomienda administrar inicialmente 1,2-1,5 mEq/kg, aunque en casos de depleción grave puede necesitarse hasta 2,5 mEq/kg47, ajustando las dosis en función de la respuesta clínica y la concentración sérica. El potasio iv no debe administrarse de forma rápida. Se consideran seguras velocidades de administración de 10-20 mEq/h, con un máximo de 40 mEq/h. Si la velocidad es superior a 10 mEq/h se recomienda usar una vía central y realizar monitorización cardíaca simultánea. La concentración de potasio en las soluciones no debe superar los 80 mEq/l cuando se van a infundir por vena periférica y los 120 mEq/l en el caso de vías centrales1.
Tratamiento de la hipomagnesemiaLa hipomagnesemia severa (magnesio < 1 mg/dl) se asocia a un déficit de magnesio corporal total de 1-2 mEq/kg. Los suplementos orales se absorben mal y ocasionan diarrea y molestias gastrointestinales. Debe realizarse tratamiento iv en pacientes sintomáticos o con hipomagnesemia grave. De forma empírica, se recomienda administrar en pacientes asintomáticos con hipomagnesemia leve-moderada 8-32 mEq de magnesio, hasta un máximo de 1 mEq/kg, y en pacientes sintomáticos con hipomagnesemia severa 32-64 mEq de magnesio, hasta un máximo de 1,5 mEq/kg48. La eliminación renal de magnesio es rápida (el 50 % de la dosis iv administrada se elimina en orina) y el equilibrio entre el espacio intra y extravascular se alcanza de forma lenta49, por ello se recomienda infundirlo a velocidad baja y monitorizar las cifras plasmáticas 12-24 h tras la reposición. Habitualmente, las dosis de hasta 6 g de sulfato magnésico (1 g de sulfato magnésico contiene 8 mEq de magnesio) se administran en 6-12 h y en 12-24 h las dosis superiores, sin sobrepasar una velocidad máxima de 1 g de sulfato de magnesio/h y una dosis máxima de 12 g1. Excepcionalmente, en casos de hipomagnesemia severa sintomática pueden llegar a administrarse 32 mEq de magnesio en 4-5 min50.
La reposición de fosfato, potasio y/o magnesio en pacientes diagnosticados de SR, y que además presenten alteración de la función renal, habrá de realizarse de forma cuidadosa. Así, en aquellos con aclaramiento de creatinina < 50 ml/min, creatinina ≥ 2 mg/dl u oligoanuria que no se sometan a terapia renal sustitutiva, se debería administrar inicialmente ≤ 50 % dosis empírica calculada de estos electrolitos1.
El tratamiento del déficit de tiamina y el manejo de la retención de sodio/sobrecarga de fluidos se ha reflejado en el apartado "Prevención".
ConclusiónEl SR de realimentación es un cuadro clínico potencialmente grave. Su prevención exige la detección de los individuos en riesgo, así como la realización del soporte nutricional de forma cuidadosa y con una monitorización adecuada. Una vez diagnosticado un paciente de SR habrá de suspenderse el soporte nutricional de forma inmediata, instaurar las medidas de apoyo necesarias y corregir las anomalías electrolíticas subyacentes.

