






Monitoring plasma levels of antiepileptic drugs for the treatment and prophylaxis of epilepsy is one of the strategies enabling clinical results to improve by reducing adverse affects and increasing effectiveness.
The objective of this article is to review the basic aspects in the monitoring of antiepileptic drugs using a consensus document prepared and endorsed by the pharmacokinetics and pharmacogenetics working group (PK.gen) of the Sociedad Española de Farmacia Hospitalaria (Spanish Society of Hospital Pharmacists).
La determinación de las concentraciones plasmáticas de antiepilépticos para el tratamiento y profilaxis de la epilepsia es una de las estrategias que permiten mejorar los resultados clínicos, reduciendo los efectos adversos y aumentando la efectividad.
El objetivo de este artículo es revisar los aspectos básicos en la determinación de los antiepilépticos mediante un documento de consenso realizado y avalado por el grupo de trabajo de farmacocinética y farmacogenética de la Sociedad Española de Farmacia Hospitalaria (PK.gen).
The main objective of pharmacokinetic monitoring of antiepileptic drugs is to optimise treatment by studying drug levels in biological matrices. Adapting individual doses is no easy task, due to the presence of factors including: a) the considerable inter-individual pharmacokinetic variability of antiepileptic drugs; b) the use of these drugs as prophylactics for long-term epileptic seizure control, and c) having no defined correlation between efficacy and a biological marker that could help with decision-making.1
It is obvious that epilepsy treatment has benefited from the determination of serum concentrations during treatments. High inter- and intra-individual variability, serum concentration-effect relationships, drug interactions, and so on, are all aspects that make personalising antiepileptic treatments through pharmacokinetic determination recommendable.2
However, the fact that these techniques have become widespread occasionally results in improper determination; the techniques are sometimes used unnecessarily in ways that may cause patient discomfort or result in incorrect dosage adjustments when the optimum serum drug level is considered to be the same as the normal range. Treatment efficiency decreases in such situations.3
The objective of this article is to review the basic aspects of antiepileptic drug determination using a consensus document prepared and endorsed by the pharmacokinetics and pharmacogenetics working group (PK.gen) of the Sociedad Española de Farmacia Hospitalaria (Spanish Society of Hospital Pharmacy).
General Remarks and Therapeutic Ranges of Different DrugsTherapeutic ranges are ranges in drug levels that are related to the probability of achieving a certain response. In this case, we refer to the probability of a range of serum levels being associated with clinical response in a high percentage of patients with a minimum incidence rate of adverse effects. Most studies consider a 50% reduction in epileptic seizure frequency to demonstrate clinical efficacy. For that reason, therapeutic ranges should be used and referred to as reference ranges. The true therapeutic range should be defined on an individual basis as the range of levels associated with the best possible response in a certain patient.3,4
Treatment recommendations should never be made based solely on serum drug levels. Clinical symptoms are extremely important in decision-making. It is important to note that the dose must not be increased for patients whose epileptic seizures are well controlled, even if serum levels of the antiepileptic drug are below the established reference limits.
Generally speaking, we must differentiate between reference ranges for traditional antiepileptics (phenobarbital, phenytoin, valproic acid, carbamazepine and primidone) and those for new antiepileptic drugs. Logically enough, there is a larger body of research on traditional antiepileptics. For new antiepileptic drugs, concentration/dose ratios that provide information about therapeutic non-compliance or previously unreported drug interactions are extremely useful.1
Studies of antiepileptic drugs in children show different pharmacokinetic behaviour from that observed in adults. Plasma clearance and the apparent distribution volume are generally high in the paediatric population. Furthermore, children present more inter-individual variability and more drug-resistant seizures.
In patients with kidney or liver failure who take antiepileptic drugs whose main elimination channel is altered, serum levels should be monitored with a view to providing safe, effective treatment.5
Reference range values used in this article were taken from recommendations made by the International League Against Epilepsy's subcommission on therapeutic drug monitoring.4
Antiepileptic Drugs. Reference RangesCarbamazepineRetrospective and observational studies recommend treating epileptic seizures, psychiatric disorders and trigeminal neuralgia with serum levels of carbamazepine in monotherapy at 4–12mg/l (17–51μmol/l). When making individual adjustments, the range should be thought of as variable, mainly owing to variations in the unbound fraction, the contribution of the active metabolite (carbamazepine 10,11-epoxide) and inter-individual variability in response to treatment.6
Carbamazepine 10,11-epoxide is metabolised through action of the epoxide hydrolase enzyme into an inactive diol metabolite. The relation between both metabolites and between carbamazepine and the metabolites provides key information in drug interaction situations.
In patients in whom carbamazepine is associated with enzyme-inducing antiepileptics, toxic symptoms may arise when carbamazepine levels are found within the reference range, or even at lower values, due to the increase in production of its active metabolite.4
Similarly, if carbamazepine is associated with lamotrigine, serum levels below those described above for monotherapy can keep a patient free from epileptic seizures.
Both in this case, and in association with enzyme-inducing antiepileptic drugs, we recommend not exceeding carbamazepine serum levels of 8mg/l (34μmol/l) due to the risk of adverse effects. Administration with quetiapine sometimes produces toxicity symptoms including drowsiness, ataxia, etc. due to epoxide hydrolase inhibition and accumulation of carbamazepine 10,11-epoxide.
EthosuximideRetrospective and observational studies show that ethosuximide reaches therapeutic efficacy in adult and paediatric patients with the absence seizures at serum levels of 40–100mg/l. However, patients with resistant epileptic seizures, including absence of seizures, require serum concentrations approaching 150mg/l. Theoretically, patients with structural lesions in the central nervous system have a greater likelihood of responding to treatment.4,7
PhenobarbitalThe therapeutic efficacy of phenobarbital in adult patients has been associated with quite a wide range of serum levels: 10–40mg/l (43–172μmol/l). Higher phenobarbital levels may be required for treating partial seizures than for treating generalised seizures.6,7
PhenytoinThe therapeutic efficacy of phenytoin in adult patients is associated with a wide range of serum levels: 10–20mg/l (40–79μmol/l), with considerable variability and efficacy margins that overlap toxicity margins.
Phenytoin has a high rate of union with serum albumin (90%). In conditions in which hypoalbuminaemia is present, such as cirrhosis of the liver or pregnancy or in critical patients, the total phenytoin level must be corrected because these conditions correspond to a larger proportion of unbound phenytoin, which is the pharmacologically active type.6
A different range of total phenytoin levels was established in newborns, 6–14mg/l (25–55μmol/l), due to the drug's lower binding rate with neonatal serum albumin.8 Similarly, the reference range in elderly patients is also lower due to increased sensitivity to phenytoin and potential changes in the unbound drug fraction.
GabapentinThe therapeutic efficacy of gabapentin is associated with a wide range of serum levels: 12–20mg/l (70–120mmol/l),2 due to the large fluctuations in serum levels associated with a short elimination half-life.
The absorption of gabapentin is dose-dependent, and therefore dosage adjustments require pharmacokinetic monitoring. It is eliminated through the kidneys, and adjusting the dose is necessary if kidney function should undergo any significant changes.1
Cimetidine decreases plasma clearance of gabapentin, and antacids (aluminium and magnesium compounds) can reduce its absorption by more than 24%.1
LamotrigineMorris et al.9 suggest a range of lamotrigine levels of 3–14mg/l9 for patients with resistant epilepsy. Other authors expand this range as far as 2.5–15mg/l (10–60μmol/l). In patients treated with lamotrigine associated with vigabatrin, the range of lamotrigine levels observed in responsive patients corresponded to a median plasma level of 7.9mg/l.10 However, no clear cut-off point has been established that would classify patients as responsive or non-responsive. We must point out that the toxicity incidence increases significantly when serum concentrations rise above 15μg/ml.
Lamotrigine has numerous interactions with enzyme inducers and inhibitors. Its interaction with valproic acid is particularly relevant because that drug is prescribed frequently and causes a significant increase in serum lamotrigine levels.
Plasma clearance of lamotrigine also increases significantly during pregnancy. In the Petrenaite et al.11 study, the dosage requirements in the third trimester of pregnancy were triple those of the patient prior to her pregnancy.
LevetiracetamMINCEP,12 the epilepsy care research group, proposed a reference range of 12–46mg/l (70–270μmol/l) for levetiracetam. The lower limit of this interval was validated by Lancelin et al.13 in a study of 69 patients carried out in 2007.
Levetiracetam is predominantly eliminated by the renal route, and for that reason, its plasma clearance is strongly dependent upon renal function and patient age. However, a small fraction of the administered dose of levetiracetam is metabolised by the liver, and for that reason, drug clearance increases when levetiracetam is used in combination with enzyme-inducing antiepileptics such as phenytoin or phenobarbital.4
OxcarbazepineOxcarbazepine is a pro-drug that quickly and almost totally metabolises its active 10-monohydroxy metabolite (MHD), also known as licarbazepine, which is responsible for its therapeutic effect. For that reason, the reference range refers to the concentrations of this metabolite. A range of levels from 13 to 35mg/l (50–140μmol/l) has been proposed for MHD monitoring, although a high degree of variability and overlapping have been observed in both the efficacy and the toxicity margins.7
The elimination processes of oxcarbazepine and its metabolite MHD are independent from cytochrome P450, and they occur through non-oxidative processes, which are ketone reduction and O-glucuronidation, respectively. For this reason, its plasma clearance is not affected by concomitant use of other enzyme inducers or inhibitors.14 However, it has been observed that inducers such as phenobarbital, phenytoin and carbamazepine reduce MHD metabolite concentration.
The tables showing MHD level/dose, proposed by Armijo et al.15 may be useful for routine oxcarbazepine monitoring.
PrimidoneThe therapeutic efficacy of primidone in adult patients is associated with a serum level range of 5–10mg/l (23–46μmol/l). Primidone is metabolised to phenobarbital, which is largely responsible for its activity. It is therefore possible to personalise primidone treatment by monitoring phenobarbital levels. The ratio of primidone concentrations to phenobarbital in monotherapy is 1:2. This proportion is higher for polytherapy and in children.7,16
TiagabineThis drug's reference range is from 20 to 200ng/ml (53–532nmol/l). Monitoring has only been shown to be useful in cases of suspected intoxication and to confirm treatment adherence. In both cases, it is important to know the elapsed time between drug administration and sample collection since the biological half-life is extremely variable, between 5 and 9h, in patients on monotherapy and between 2 and 4h when administered with enzyme inducers.1,17
TopiramateThe generally accepted reference range is 5–20mg/l (15–60μmol/l). Numerous studies have identified properly managed epilepsy with good drug tolerance at values between 2 and 10mg/l. Levels higher than 20mg/l are associated with adverse effects and encephalopathy.1,3
Valproic AcidThe accepted reference range for valproic acid is 50–100mg/l (347–693μmol/l). Serum levels above 175mg/l are associated with a high risk of neurotoxicity.
There is little correlation between the dose that is administered and serum valproic acid levels due to its high plasma protein binding. This process is saturable, even at therapeutic levels. When this occurs, changes in the unbound fraction of the antiepileptic are not proportional to the dosage increase. For patients with hypoalbuminaemia, Hermida and Tutor18 propose using a table to correct serum levels of unbound valproic acid according to the patient's albumin levels, thereby preventing potential toxic effects.
In elderly patients, it is important to recall that the unbound fraction may be higher due to the decrease in plasma proteins. On the contrary, children need higher doses than those administered to adults in order to achieve similar serum levels.
Monitoring unbound concentrations is recommended for pregnant patients because changes in maternal proteins result in a decrease in the total valproic acid values without there being any significant changes to the unbound concentrations.19
VigabatrinVigabatrin binds irreversibly to GABA-transaminase, which causes the lack of correlation between drug concentration and drug effect, owing to the time required for enzyme regeneration. It was not possible to establish a correlation between concentration and therapeutic effect. However, given normal doses, serum levels range between 0.8 and 36mg/l (6–279μmol/l), with a proportional relationship between the administered dose and the serum concentration obtained.1,3
ZonisamideThe reference range for zonisamide is established at 10–40mg/l (47–188μmol/l). However, in published clinical studies, most patients experience adverse effects at levels higher than 30mg/l.1,17
Practical Aspects of Pharmacokinetic MonitoringSituations in Which Therapeutic Monitoring is RecommendedAntiepileptic monitoring should only be undertaken as an aid in making drug treatment decisions. There is no rationale for systematic monitoring.
The indications for pharmacokinetic monitoring, for both new antiepileptics and traditional antiepileptics, are as follows:3,4,7,20
- 1.
When it becomes necessary for clinical reasons to reduce the risk of seizure recidivism by adjusting the patient's serum levels within the applicable reference range, or in the upper limits of that range.
- 2.
Patients receiving antiepileptics whose pharmacokinetic behaviour is saturable, such as phenytoin, and in cases in which it is difficult to predict serum levels based on the initially prescribed dose.
- 3.
Suspected toxicity, or where there is an inconclusive differential diagnosis of signs and symptoms stemming from toxicity due to antiepileptic levels.
- 4.
To define the patient's individual therapeutic range. In patients whose epilepsy has been controlled and maintained over a sufficiently long time span, it is better to monitor antiepileptic serum levels twice, over the span of several months, rather than just once. This allows us to estimate measurement variability.
- 5.
Poorly controlled epilepsy. This allows us to identify the cause of the relapse or any changes that must be made to the dosage.
- 6.
Pregnancy. During pregnancy, physiological changes affect antiepileptic pharmacokinetics and drug levels in both the woman and the foetus. For patients whose condition is well controlled, we recommend monitoring serum levels for the drug once every 3months. In women with complicated epilepsy, or those taking lamotrigine or oxcarbazepine, we recommend increasing monitoring to once per month.
- 7.
Population groups and physiopathological stages thought to experience changes in the pharmacokinetic behaviour of these drugs, including paediatric and geriatric patients, patients with bariatric surgery, kidney or liver failure, infectious diseases, large burns, critical patients, etc.
- 8.
If there are changes in the drug format or content.
- 9.
Suspected drug interaction.
In general, the optimum extraction time for monitoring antiepileptic treatments that are administered orally is just before the morning dose. The resulting level is alternately known as the trough, baseline, minimum or pre-dose level.
This sample strategy is particularly important for drugs with a short elimination half-life. For drugs whose elimination half-life is greater than 24h, the time during the dosing interval when the sample is extracted is less critical.
However, we must state that the optimum sample extraction time depends on the clinical situation and the reason for ordering drug monitoring.
Emergency Situations- –
Suspected intoxication or adverse effect depending on drug level. Blood samples must be taken as early as possible in order to determine the most appropriate treatment (haemoperfusion, hydration and alkalinisation, administration of activated charcoal, etc.). The sample time with regard to time of last dose does not limit monitoring, although we must gather all necessary information with respect to the elapsed time between drug administration and extraction of the sample in order to interpret the result correctly.
- –
Suspected treatment non-compliance or underdose. The sample must be taken once the drug distribution equilibrium has been reached. Otherwise, the correlation between plasma level and pharmacodynamic response may already be imbalanced, which could lead to improper dosage adjustments. If phenytoin is administered intravenously, the post-dose sample must be taken at least 2h after administration of the last dose.
- –
Measurements should be made before administering the first morning dose, except for phenobarbital (ideally, pre-dose level) which has such a long elimination half-life that it only presents mild fluctuations in level. On the other hand, while phenytoin also has a long elimination half-life, the time needed to reach the maximum level fluctuates considerably owing to both the pharmaceutical format and the administered doses. This makes taking morning baseline measurements recommendable.
- –
In either case, when comparative measurements are taken, it is important that sample times be coherent with respect to the last dose. In the case of valproic acid, levels may vary nearly 100% during the dosing interval, and we must be particularly careful to always take samples at the same time with respect to the dose time.
- –
Likewise, measurements must always be taken at the same time of day in order to avoid circadian influences (for valproic acid and carbamazepine).
Monitoring frequency depends on the clinical situation which made it necessary to monitor levels the first time, and on response to the initial treatment.
Emergency Situations- –
After discontinuing treatment due to toxicity or adverse effects in order to assess risk of toxicity.
- –
After the interval estimated to be sufficient in order for levels to reach therapeutic values according to the elimination half-life of each drug, or preferably, the patient's pharmacokinetic history, if available.
- –
At 72–96h after starting treatment, even if steady state has not yet been reached, in order to rule out sub-therapeutic or toxic drug levels. If the dosage is changed after taking the first measurement, levels will be monitored once more after 72–96h or after having reached steady state.
Carbamazepine has an enzyme self-induction process that begins 24h after starting treatment and lasts between 1 and 5weeks. We should therefore be mindful of the influence of this effect on both levels of carbamazepine itself and on any associated drugs in the case of polytherapy.
Monitoring During Steady State- –
First-order elimination drugs reach steady state after approximately 5 elimination half-lives. Table 1 shows the time required by different antiepileptic drugs in order to reach steady state.
Table 1.Elimination Half-life of Antiepileptic Drugs and Time to Reach Steady-state.
Antiepileptic Steady State, Days Elimination Half-life, h Carbamazepine 2–6 Single dose: 25–65Self–induction: 16 Phenytoina 4–24Lead dose: 48–96h Low dose: 6–12High dose: 12–60 Phenobarbital 10–25 90–100 Primidone 2–4 9–22 Valproic acid 2–4Lead dose: 24h 15 Ethosuximide 5–15 30–60 Clonazepam 6 10–30 Gabapentin 2 5–9 Lamotrigine 5–6 15–60 Tiagabine 1–2 5–8 Topiramate 4–6 12–30 Vigabatrin 2 5–8 Levetiracetam 5 7 Oxcarbazepine 1 1–2 Felbamate 4 14–23 - –
For zero-order elimination drugs, the elimination half-life increases when the metabolism saturation point is reached. Monitoring is particularly recommended when treatment response is correct and prior serum concentrations are within the reference range or approaching the lower limit.
- –
We recommend that check-ups be performed: 1) yearly in adult patients with good compliance, no treatment modifications in the past year and a well-managed condition; 2) every 6months in paediatric patients with good compliance, no treatment modifications in the past 6months and a well therapeutically managed condition; and 3) quarterly or more often whenever treatment is modified (dose or interval), when drugs with the potential to cause interactions are included, in non-compliant children, and when there are changes in liver, heart or gastrointestinal function or in case of pregnancy, etc.23
Systematic determination of antiepileptic levels in order to keep them within the reference range, regardless of the patient's clinical solution, is a debatable practice because it delivers scant benefits, results in patient discomfort and places an increased burden on the healthcare system. Some authors recommend monitoring only when there is a clear clinical rationale behind it. However, other publications recommend monitoring antiepileptic levels, particularly as a guideline for personalising treatment in patients with infrequent epileptic seizures.24 The same authors also consider that it is inappropriate to monitor patients undergoing chronic carbamazepine or valproic acid treatment and whose dosage has changed because their kinetics follow a predictable slope.
In outpatients, monitoring tends to be done for the first time 4weeks after administration of the full drug dose because steady state will have been achieved by that time. In hospitalised patients, monitoring is completed for the first time once steady state is reached, but if a shock dose is administered or if there is an emergency, levels may be measured before that time, and dosage may be adjusted according to patient characteristics and an estimate of the drug level at steady state.
General Recommendations for Creating the ReportThe result of a monitoring request is a pharmacokinetic report that presents the following information:
- –
Serum levels observed after analytical monitoring, and their correlation with the reference interval.
- –
Achievement of steady state.
- –
Pharmacotherapeutic recommendations, including, if necessary, loading dose, time to begin new dose, etc.
- –
Need for new tests.
- –
Identification of any factors that may have influenced the serum level results (interactions, inappropriate sample times, treatment non-compliance, etc.).
- –
Pharmacokinetic parameters (optional).
Information on the patient's clinical situation, obtained through communicating with the lead doctor and nursing staff and consulting the medical history, must be available in order for the report to be drawn up. Serum level results and any recommendations that are made must be documented in the patient's medical history, along with information used to prepare the report.
The pharmacokinetic report must be made available, whether in oral or written format, to the prescribing doctor before the next dose can be given to a hospitalised patient. For outpatients, the report must be ready before the patient's next visit to the lead doctor. When potentially toxic or ineffective levels are identified or suspected, the maximum recommended time is 4h for both outpatients and hospitalised patients.
Analytical Aspects of Antiepileptic MeasurementSample TypeSerum is the recommended biological matrix for quantifying antiepileptic levels. Anticoagulants used in order to obtain plasma may loosen drug molecules bonded to proteins.7 Sodium heparin may activate lipoprotein-lipases in such a way that when fatty acid levels rise, the drug/protein bond will be disrupted. For example, sodium citrate or oxalate decreases the total levels of phenytoin and valproic acid. This is why using plasma instead of serum would require us to check for interactions. We also recommend caution when using collection tubes that contain gels, as these may cause adsorption phenomena.
Analytical Techniques for Quantitative Measurement of Antiepileptic Drugs Subject to Clinical MonitoringIn general, immunoanalysis techniques including fluorescence polarisation immunoassays (FPIA), photometric immunoassays (EMIT or CEDIA), turbidimetric assays (PETINIA, QMS) and chemiluminescent assays (CMIA) are the most common in clinical practice due to their ease of use and the fact that they run on large-scale analytical platforms that allow results to be received quickly. Chromatographic techniques, such as gas chromatography and high-performance liquid chromatography (HPLC), are more versatile, sensitive and specific, and this makes them a very interesting alternative to immunoassays. However, these chromatographic techniques still have their limitations, such as high cost and the need for highly qualified personnel. In addition, it is usually necessary to treat biological samples before they can be injected in to the chromatograph. For example, HPLC methods for antiepileptic monitoring include prior extraction of the samples either by precipitation, liquid-liquid extraction or a solid phase extraction using extraction cartridges. A preliminary step involving evaporation and reconstitution of resulting residue is also common.
Martinavarro-Domínguez et al.25 developed a micellar HPLC method for carbamazepine, phenobarbital and phenytoin testing with results that were slightly inferior to those obtained using FPIA, although there were no significant differences where clinical interpretation was concerned.
Given that it is very common for patients to be treated with several different antiepileptic drugs, monitoring them simultaneously is an interesting prospect. To date, only a few analytical chromatography methods that make multiple, simultaneous measurements have been described in print. We can, however, highlight works by Bugamelli et al.26 and Vermeij et al.27 in which researchers simultaneously measured lamotrigine, oxcarbazepine, carbamazepine, phenobarbital, primidone, phenytoin and active carbamazepine and oxcarbazepine metabolites.
The incorporation of mass spectrometers has revolutionised drug analysis and chromatography. This leads us to picture chromatography with mass spectrometry as the ideal technique to be implemented in the clinical laboratory in the near future. However, the need to hire qualified personnel and the high cost of the technology are significant drawbacks to its being used in clinical practice. This is the main reason why HPLC and immunoassay methods are still the reference techniques. Most of the new antiepileptic drugs (felbamate, gabapentin, lamotrigine, levetiracetam, oxcarbazepine, tiagabine, topiramate, vigabatrin and zonisamide) are monitored using conventional HPLC with UV detection, with a significant and progressive increase in mass spectrometer use. Unlike with HPLC, chromatography measurements by means of a mass spectrometer depend on the mass of the ionic molecule or ionic fragment of the drug instead of the drug's UV absorption properties. This is why that technique is a good alternative for measuring compounds with a low UV absorption capacity. In addition, given the technique's greater specificity, a drug may be easily detectable by its molecular ion, even if it is not completely free from other co-eluting compounds or interferences. Therefore, treatment of the sample prior to chromatography would not be as decisive as it is in the case of chromatography using a UV detector. Lamotrigine, topiramate and zonisamide are 3 antiepileptic drugs for which microparticle enzyme immunoassay (MEIA) methods have been developed; FPIA may also be used for topiramate. Results obtained for all 3 microparticle techniques are similar to those delivered by HPLC.
Other less commonly used techniques include capillary electrophoresis or ion-selective membrane electrodes. The capillary electrophoresis methods that have been described deliver results similar to those obtained with chromatographic or immunoassay methods of reference.28 Gupta et al.29 developed selective potentiometric electrodes for measuring lamotrigine, felbamate and primidone; results indicate that these electrodes are precise and safe for measuring levels of these drugs.
Tsanaclis et al.30 evaluated the precision of different assays for antiepileptic drugs in the most commonly used automated analysers.30 Most of the analytical methods delivered comparable levels with coefficients of variation consistently below 10% and an accuracy of about 7%.
Regarding chromatographic techniques, in all of the analytical methods that were studied, results for precision were very similar to those obtained using the immunoassay method. The small differences in accuracy with respect to immunoassay results may be due to a lack of specificity and the cross-reactivity observed for some immunoassay tests, such as the FPIA test for carbamazepine, in which accuracy fluctuates between 10.7% and 15.5%. Despite the positive deviation presented by the cross-reactivity, which arises because of the epoxide metabolite, the mean deviation from the mean theoretical value is paradoxically negative (−8.26%). This seems to be related to the non-human matrix used in reagent preparation.
Table 2 shows the methods used in antiepileptic analysis, along with the equipment used to make the measurements.
Analytical and Instrumental Techniques Used to Determine Antiepileptic Levels in Biological Fluids.
| Drug | Analytical Technique | Instrument® |
| CarbamazepineEthosuximidePhenytoinPhenobarbitalFelbamateGabapentinLamotrigineLevetiracetamOxcarbazepinePrimidoneTiagabineTopiramateValproic acidVigabatrinZonisamide | FPIA, EMIT, CEDIA, GLC, LC–MS, CEFPIA, EMIT, CE, HPLC, GLCFPIA, EMIT, HPLC, GLC, CEDIAHPLC, GLC, LC–MS, FPIA, EMIT, CEDIA, HPLC, GLCHPLC, GC–MS, LC–MSHPLC, QMS, CEHPLC, GCHPLC, GLCFPIA, EMIT, HPLC, GLCHPLC, GC–MSFPIA, HPLC, GC, GLC-NPD, LC–MS, QMSFPIA, EMIT, CEDIA, GLC, HPLCHPLCHPLC, QMS, CE | Architect, Aeroset, Axsym, TDX, ViVa E, Dimension, Olympus AU 400, HPLCTDX, ViVa E, Dimension, HPLC, GLCArchitect, Aeroset, Axsym, TDX, ViVa E, Dimension, Olympus AU 400, HPLC, GLCArchitect, Aeroset, Axsym, TDX, ViVa E, Dimension, Olympus AU 400, HPLC, GLCHPLC, GLCHPLC, GC–MS, LC–MSHPLC, Olympus AU 400, Hitachi, CDx90HPLC, GCHPLC, GCTDX, ViVa E, Dimension, HPLC, GLCHPLC, GC–MSTDX, HPLC, GC, LC–MS, Olympus AU 400, Hitachi, CDx90Architect, Aeroset, Axsym, TDX, ViVa E, Dimension, Olympus AU 400, HPLC, GLCHPLCHPLC, Olympus AU 400, Hitachi, CDx90 |
CE, capillary electrophoresis; CEDIA, cloned enzyme donor immunoassay; CMIA, chemiluminescent microparticle immunoassay; EMIT, enzyme-multiplied immunoassay technique; FPIA, fluorescence polarisation immunoassay; GC, gas chromatography; GC–MS, gas chromatography–mass spectrometry; GLC, gas-liquid chromatography; GLC/NPD, gas-liquid chromatography with nitrogen-phosphorus detection; HPLC, high-performance liquid chromatography; LC–MS, liquid chromatography–mass spectrometry.
Antiepileptic drug levels can also be measured in spinal fluid, tears and saliva, and except for gabapentin, their levels in these fluids are similar to the plasma unbound fraction. However, extracting samples of these fluids is not convenient for systematic monitoring due to the difficulty of the collection process, except in the case of saliva.31
Assessing drug levels in saliva is a more complicated process. Transfer of the drug to saliva depends on its physical and chemical properties, including molecular size, liposolubility, pKa values and protein binding.
Protein concentration in saliva is low compared to that in plasma. It is therefore considered to be insignificant and assumed to be the same as that of plasma water, except that there are differences between saliva and plasma pH that can create differences in the unbound concentration of the non-ionised drug.31
Antiepileptics that are not ionised in saliva's pH conditions may be monitored in saliva. The effect of pH holds little importance for phenytoin (pKa=9.2) and carbamazepine (pKa>12), which are largely unaffected by changes in pH that result from administering salivation stimulants. For phenobarbital (pKa=7.2), we propose correcting the saliva measurement for the pH value by using the Henderson–Hasselbalch equation22 in order to give a more accurate estimate of the drug plasma level. Other authors have found excellent correlations between saliva and plasma levels with no need to correct for plasma pH.32
Under standardised, controlled conditions, saliva can be used as an alternative matrix for measuring carbamazepine, phenytoin, primidone and ethosuximide since it has been shown to correlate well with the plasma unbound fraction for these drugs. In contrast, results of saliva testing for valproic acid and phenobarbital are a matter of debate.33,34
Finding Unbound FractionSystematic monitoring of the unbound fraction is not normally necessary for antiepileptic drugs. Drugs considered to be candidates for determining unbound fraction due to their clinical utility are those whose plasma protein-bound fraction is greater than 80%.20 Here, a relatively small decrease in the level of protein available to bind with the drug may produce a significant increase in the unbound drug concentration. Likewise, the unbound fraction may change when the extension of the protein bond changes as a result of alterations in drug level, availability or competition and affinity for protein binding sites.
Antiepileptic agents considered to be candidates for unbound fraction measurement are phenytoin, valproic acid and carbamazepine, even though the degree of protein binding does not reach 80% in the case of the latter. Tiagabine meets these criteria, but more studies are needed in order to establish its interval of reference.
The clinical usefulness of monitoring the unbound fraction of antiepileptics has been widely documented in the literature. However, some results show a contradictory correlation between the unbound fraction of the drug and its pharmacological effect.20 We have observed a high level of inter-individual variability in the unbound fraction of these drugs among patients with severe renal failure and liver disease. Malnourished children and newborns, particularly those with hyperbilirubinaemia, present decreased drug-to-protein binding.
Valproic acid is characterised by high plasma protein binding, primarily to albumin, and the fact that its bonds are saturable at therapeutic doses (75mg/l). In situations in which its intrinsic plasma clearance remains unchanged, but plasma protein binding is affected, we recommend measuring the unbound fraction. The percentage of unbound valproic acid may rise from 5%, with total levels at 10–60mg/l, to 10% when levels are at 75–100mg/l, to even 20%–30% when total levels are at 145–160mg/l. Increases in the percentage of unbound drug of more than 50% have been recorded.18
Valproic acid presents a low rate of renal extraction, with total plasma clearance depending on the unbound fraction and on intrinsic liver clearance. A decrease in albumin binding will increase the free fraction which is pharmacologically active, but it increases the free fraction available for elimination as well, which will also augment total valproic acid clearance. This results in a decreased total plasma level, even if free valproic acid levels should remain the same or increase. A dosage increase would augment the unbound fraction, which would multiply the risk of toxicity. It is therefore important to detect situations in which plasma protein binding displacement or decreased protein binding occur, including kidney or liver failure, hyperbilirubinaemia, excess of free fatty acids in plasma, uraemia, drug interactions, etc.35,36
This is the case with salicylic acid, which competes with valproic acid for albumin-binding sites. In vitro studies have identified increases in the unbound fraction of valproic acid in HIV positive patients (HIV+) as a result of hypoalbuminaemia and possible interactions between valproic acid and antiretroviral treatment.20 We also recommend measuring the unbound fraction of valproic acid in patients whose disease is not well controlled after a dosage increase.37
Some authors have developed indirect methods for estimating the unbound fraction (KODAMA). Hermida et al.13 propose a mathematical model to normalise the total level of valproic acid in patients with hypoalbuminaemia based on the following equation: CN=αH·CH/6.5, where CN is the total normalised level of valproic acid, αH is the percentage of unbound fraction calculated for the patient, or where unavailable, the percentage these authors suggest for different serum albumin values, and CH is the total measured level of valproic acid. This methodology may be useful for personalising doses in patients with hypoalbuminaemia when the free fraction is unavailable.18
Nevertheless, we recommend measuring the unbound fraction directly. To this end, using ultrafiltration to separate the unbound fraction from the plasma protein-bound fraction and determining the antiepileptic level in the ultrafiltered sample is sufficient.38
Phenytoin's plasma protein binding fraction is 90%, and it binds mainly to albumin, but unlike the case of valproic acid, the bond is not saturable. The reference range is 0.8–2.1mg/l.
The situations that affect the percentage of phenytoin plasma protein binding are similar to those listed for valproic acid. Phenytoin binding to albumin may be significantly affected in the presence of severe uraemia and in situations involving structural modifications at albumin-binding sites. In cases of both hypercholesterolaemia and mixed hyperlipidaemias, the increase in free fatty acids displaces phenytoin from its albumin bond, and its unbound fraction increases. In patients with eclampsia, the unbound fraction of phenytoin is abnormally high, but neither the total concentration nor the unbound concentration is a good predictor of patient response. Phenytoin binding to proteins may also be affected by the presence of other acid medications such as valproic acid, salicylates, phenylbutazone and sulphonylureas. Lastly, we observed that in HIV-positive patients, the unbound phenytoin concentration is high as a result of hypoalbuminaemia or because of phenytoin interaction with the multiple treatments that these patients receive.20
In a study of 139 patients, the unbound fraction of phenytoin fluctuated between 6.8% and 35.3% of the total plasma level. The clinical situations responsible for this variability were hypoalbuminaemia, drug interactions, uraemia, pregnancy and very young or old ages. The authors concluded that total plasma level of phenytoin was less reliable than the free phenytoin level as an indicator of effectiveness. Recently, Iwamoto et al.39 reiterated the need for monitoring the unbound fraction of phenytoin in patients undergoing monotherapy.
When patients suffer hypoalbuminaemia and there is no available analytical measurement of the phenytoin unbound fraction, measured in a procedure similar to that for valproic acid, the total plasma phenytoin content can be normalised or corrected by using the Sheiner–Tozer equation:
where Cobserved is the total phenytoin concentration observed after the analytical measurement and Alb. is the patient's albumin concentration expressed in g/dl.37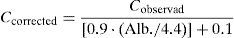
Similarly, in patients with hypoalbuminaemia who undergo kidney replacement therapy techniques, the equation shown above can be modified in order to normalise total plasma phenytoin levels by replacing the value of 0.9 with 0.43.
The plasma protein binding of carbamazepine is 70%–80%, with a rate of 50% for its main active metabolite, carbamazepine 10,11-epoxide. Carbamazepine presents lower variability in plasma protein binding than valproic acid or phenytoin. However, determining the unbound fraction of carbamazepine in patients receiving monotherapy is less useful because there is a larger overlap between potentially therapeutic and potentially toxic levels of unbound carbamazepine (interval of reference for unbound fraction of carbamazepine: 0.9–2.8mg/l).34,20 However, monitoring is justified in patients with uraemia or liver disease or in pregnant women because they may experience unexpected increases in the unbound fraction of carbamazepine.
To summarise, monitoring the unbound fraction of valproic acid, phenytoin and carbamazepine is indicated in the following situations20,35:
- –
Total serum levels within the reference range in the presence of signs or symptoms of toxicity or lack of proper patient response.
- –
Patients with uraemia or severe kidney failure.
- –
Patients with liver disease.
- –
Patients with hypoalbuminaemia or albumin levels <2.5g/dl.
- –
Concomitant medication with high plasma protein-binding capacity that may compete with the antiepileptic for plasma protein bonds.
- –
Patients with hyperlipidaemia.
- –
Patients infected with HIV.
Traditional antiepileptic drugs are normally eliminated through metabolism by cytochrome P450 and a number of transporters. This is why they are subject to drug interactions, whether with other antiepileptics or with the rest of the medications taken by the patient.40–42Table 3 shows the different isoforms of cytochrome P450 responsible for metabolising traditional antiepileptics, in addition to enzyme isoforms that may be induced or inhibited by these drugs, which would cause changes in the antiepileptic serum levels. Tables 4–6 list the main clinically relevant drug interactions.
Cytochrome P450 Enzymes Involved in Antiepileptic Metabolism and the Inducer/Inhibitor Effect of the Drug on Cytochrome P450.
| Metabolism | Induction | Inhibition | |
| Carbamazepine | CYP3A4, CYP2C8 | CYP2C9, CYP3A4 | |
| Ethosuximide | CYP3A4, CYP2E, CYP2B, CYP2C | ||
| Phenytoin | CYP2C9, CYP2C19 | CYP2C9, CYP3A4, UGT | CYP2C9 |
| Phenobarbital | CYP2C9, CYP2C19, CYP2E1 | CYP2C9, CYP3A4, UGT | |
| Lamotrigine | UGT | UGT | |
| Oxcarbazepine | CYP3A4, CYP3A5 | CYP2C19 | |
| Primidone | CYP2C9, CYP3A4, UGT | ||
| Tiagabine | CYP3A4 | ||
| Topiramate | β-Oxidation | CYP2C19 | |
| Valproic acid | CYP2C9, CYP2C19, CYP2A6β-Oxidation, UGT | CYP2C9, UGT | |
| Zonisamide | CYP3A |
UGT, uridine diphosphate glucuronosyltransferase.
Interactions Between Antiepileptics. Effect on Plasma Levels.
| PB | PHT | PRM | ESM | CBZ | VPA | LTG | TPM | TGB | |
| PB | SI | ↑↑/↓↓PHT | – | ↓ESM | ↓↓CBZ | ↓↓VPA | ↓↓LTG | ↓↓TPM | ↓↓TGB |
| PHT | ↑↑PB | SI | ↑/↓ PRM | ↓ESM | ↓↓CBZ | ↓↓VPA | ↓↓LTG | ↓↓TPM | ↓↓TGB |
| PRM | – | ↑↑/↓↓PHT | SI? | ↓ESM | ↓↓CBZ | ↓↓VPA | ↓↓LTG | ↓↓TPM | ↓↓TGB |
| ESM | 0 | ↑PHT | 0 | – | 0 | ↓↓VPA | 0 | 0 | 0 |
| CBZ | 0 | ↑↑/↓↓PHT | ↓ PRM | ↓ESM | SI | ↓↓VPA | ↓↓LTG | ↓↓TPM | ↓↓TGB |
| VPA | ↑↑PB | ↑↑/↓↓PHT | ↑ PRM | ↑/↓ ESM | ↑↑ CBZ E | – | ↑↑LTG | 0 | 0 |
| LTG | 0 | 0 | 0 | 0 | ↑ CBZ E | 0 | SI | ? | ? |
| TPM | 0 | ↑↑PHT | 0 | 0 | 0 | ↓↓VPA | ? | – | ? |
| TGB | 0 | 0 | 0 | ? | 0 | ↓VPA | ? | ? | ? |
?, interaction unknown or not researched; ↑, may increase plasma level somewhat; ↑↑, may increase plasma level considerably; ↓, may decrease plasma level somewhat; ↓↓, may decrease plasma level considerably; 0, no interaction; SI, self-induction; CBZ E, carbamazepine epoxide; CBZ, carbamazepine; ESM, ethosuximide; LTG, lamotrigine; PB, phenobarbital; PHT, phenytoin; PRM, primidone; TGB, tiagabine; TPM, topiramate; VPA, valproic acid.
Inducer or Inhibitor Effect of Antiepileptics on Other Drugs.
| Antiepileptic | Drug |
| Enzyme inducers | |
| • Phenytoin• Phenobarbital• Carbamazepine• Primidone• Oxcarbazepine | Antibiotics: praziquantel, albendazole, chloramphenicola (PB only), doxycycline, metronidazoleAntifungals: griseofulvin, itraconazoleAntivirals: nevirapine, efavirenz, delavirdine, indinavir, ritonavir, saquinavirAntineoplastic drugs: cyclophosphamide, ifosfamide, busulfan, teniposide, paclitaxel, methotrexate, vinca alkaloidsAntiarrhythmics: disopyramideb, mexiletine, quinidine, amiodaronebAntihypertensives: propranolol, metoprolol, alprenolol, nifedipine, felodipine, nimodipine, nisoldipine, verapamil, losartancCardiotonic drugs: digoxindHypolipidaemic drugs: atorvastatin, lovastatin, simvastatin, fluvastatinOral anticoagulants: dicoumarol, warfarinImmunosuppressants: ciclosporin, tacrolimus, sirolimusAntidepressants: amitriptyline, nortriptyline, imipramine, desipramine, clomipramine, doxepin, mianserin, nomifensine, bupropionf, nefazodone, citalopram, paroxetineAntipsychotics: haloperidol, chlorpromazine, thioridazine, clozapine, olanzapine, risperidone, quetiapine, ziprasidoneBenzodiazepines: clobazam, diazepam, nordazepam, clonazepam, alprazolam, midazolamgCorticosteroids: hydrocortisone, cortisol, dexamethasone, prednisone, prednisolone, methylprednisoloneOral contraceptiveshOther: theophylline, fentanyl, methadone, pethidine, paracetamol, thyroxine, and neuromuscular blockers |
| Enzyme inhibitors | |
| • Valproic acid | Antihypertensives: nimodipineCytostatic drugs: nitrosourea-cisplatiniAntivirals: nevirapine, efavirenz, delavirdine, indinavir, ritonavir, saquinavir, zidovudineHypolipidaemic drugsj: atorvastatin, lovastatin, simvastatin, fluvastatinImmunosuppressants: ciclosporinkTricyclic antidepressants: amitriptyline, nortriptyline, clomipramine, paroxetineAntipsychotics: haloperidol, chlorpromazine, thioridazine, olanzapine, quetiapine, ziprasidone, diazepam, lorazepam |
The formation of active metabolites of disopyramide and amiodarone makes the effect of this drug interaction more unpredictable. The magnitude of the drug's interaction with dihydropyrimidine calcium channel blockers means that it is not recommended for use with enzyme inducers.
The clinical significance with bupropion is not well-defined due to the presence of an active metabolite.
Low clinical relevance due to benzodiazapines’ wide therapeutic interval, except for oral midazolam.
Drugs that Increase (↑) or Decrease (↓) Serum Antiepileptic Levels.
| Drug | Antiepileptic |
| Antibiotics | |
| - Isoniazid | ↑ CBZ, ESM, PB, VPA, ESM |
| - Rifampicin | ↓ PHT, CBZ, VPA, ESM, LTG |
| - Carbapenems | ↓ VPA |
| - Macrolides, metronidazole | ↑ CBZ |
| - Chloramphenicol | ↑ PHT, PB |
| - Sulphonamides | ↑PHT |
| Antifungals | |
| - Ketoconazole, fluconazole | ↑ CBZ |
| - Miconazole, fluconazole | ↑ PHT |
| Antivirals | |
| - Indinavir, ritonavir, delavirdine | ↑ CBZ, ESM, TGB, ZNS |
| - Nevirapine, efavirenz | ↓ CBZ, ESM, TGB, ZNS |
| Antineoplastic drugs | |
| - Fluorouracil, UFT, tegafur, tamoxifen, doxifluridine | ↑ PHT |
| - Vinblastine, MTX, bleomycin, carmustine±cisplatinetoposide | ↓ PHT |
| - Cis-pt | ↓ CBZ, VPA |
| - MTX | ↓VPA |
| Cardiovascular drugs | |
| - Amiodarone | ↑ PHT |
| - Diltiazem, ticlopidine | ↑ PHT, CBZ |
| - Verapamil | ↑ CBZ |
| - Dicoumarol | ↑ PHT, PB |
| Gastrointestinal drugs | |
| - Antacids | ↓ PB, PHT, CBZ, GABA |
| - Sucralfate | ↓ PHT |
| - Cimetidine, omeprazole | ↑ PHT, CBZ |
| Immunosuppressants | |
| - Tacrolimus | ↑ PHT |
| Psychotropic drugs | |
| - Fluoxetine, fluvoxamine, trazodone, viloxazine | ↑ PHT, CBZ |
| - Sertraline | ↑ PHT, LTG, VPA |
| - Imipramine | ↑ PHT |
| - Nefazodone | ↑ CBZ |
| Hormones | |
| - Danazol | ↑ CBZ |
| - Oral contraceptives | ↓ LTG |
| Others | |
| - Dextropropoxyphene, alopurinol, tolbutamide, chlorphenamine, disulfiram, phenylbutazone | ↑ PHT, CBZ PB |
CBZ, carbamazepine; ESM, ethosuximide; GABA, gabapentin; LTG, lamotrigine; PB, phenobarbital; PHT, phenytoin; PRM, primidone; TGB, tiagabine; TPM, topiramate; VPA, valproic acid; ZNS, zonisamide.
There is currently enormous interest in determining the influence of genetic polymorphism on pharmacokinetics with a view to optimising drug therapy. However, data regarding its clinical relevance for drug availability are contradictory. Williams et al.43 evaluated the reasons behind these discrepancies and concluded that most studies did not include a large enough sample size for the magnitude of the measured effect to reflect the true influence of polymorphism.
Out of all of the enzyme isoforms that intervene in antiepileptic drug metabolism and that have allelic variants with a proven, significant influence on drug availability, CYP2CN and CYP2C19 are key players. The allelic variants in question for these 2 isoforms are *2 and *3, at least in the Caucasian population. The study by Klotz analyses these aspects.43
PB is partially metabolised by CYP2C19, but the limited percentage of the drug eliminated by this route means that the effect had by different polymorphisms of this isoform does not have a significant impact on clinical management of the antiepileptic.
Both isoforms intervene in phenytoin metabolism, with CYP2C9 being predominant. The role of CYP2C19 is more relevant when levels are high. There is a lack of consensus as to the influence of polymorphisms on phenytoin pharmacokinetics, which is not only due to design limitations in the studies, as mentioned above, but also due to considerable variability among different ethnic groups. Klotz described the case of an African-American patient with an allelic variant (CYP2C9*6) that is extremely infrequent in that ethnic group (0.6%) and completely absent among Caucasians. The patient's exposure to phenytoin was 5.8 times higher than that in patients possessing wild-type alleles.43
Another study observed that patients possessing the variant CYP2C9*3 may require a 13% decrease in dosage. This is the only variant with clinical repercussions. With this in mind, both genetic variants must be evaluated simultaneously in order to gather better-adjusted results. Nevertheless, pharmacokinetic monitoring of phenytoin and estimates of personal pharmacokinetic parameters already takes pharmacogenetic variability into account, allowing us to design safe and effective drug therapy dosing schemes.
The main routes of elimination for valproic acid are glucuronidation, beta-oxidation and omega-oxidation. The CYP2C9 acts in the formation of a metabolite called 4-en-valproate that is related to liver toxicity caused by the antiepileptic drug. To date, however, completed studies have failed to show an association between the possible genetic variants of CYP2C9 and liver toxicity incidence rate, which may be due to the action of another mechanism independent from CYP.
ConclusionsMonitoring serum levels of antiepileptic drugs plays an important role in optimising epilepsy treatment, which is mainly due to the considerable pharmacokinetic variability of these drugs, their clinical use as prophylactic treatments, and their narrow therapeutic margin. This article reviews the main recommendations and practical aspects for proper monitoring of these drugs. The indicated reference values define the serum levels at which most patients respond correctly. They are taken from recommendations issued by the International League Against Epilepsy's subcommission for therapeutic drug monitoring. Nevertheless, the correlation between drug level and response is not always stable over time for a specific patient. Changes in a patient's state, concomitant diseases, changes in protein binding, interactions with other drugs, etc. may modify the correlation, and therefore monitoring antiepileptic serum levels provides important information for dosage adjustment.
Conflict of InterestThe authors have no conflicts of interest to declare.
Please cite this article as: Aldaz A, et al. Monitorización farmacocinética de antiepilépticos. Farm Hosp. 2011;35:326–39.

