

To compare the measures taken by the European Union, Switzerland and the United Kingdom to ensure the continuity of the medical devices market, complying with the requirements of Regulation 2017/745.
MethodTo carry out this work, a review was made of the official websites of the European Commission, the Spanish Agency for Medicines and Health Products, the Swiss Agency for Therapeutic Products and the Medicines and Healthcare Products Regulatory Agency of the United Kingdom. Bibliographic searches were also conducted on Pubmed and the internet (Google), using terms such as “withdrawal of the Mutual Recognition Agreement of Swiss European Union medical device conformity certificates, new UK medical device regulation”, for a period extending from January 2020 to December 2021.
ResultsAs a result of the disappearance of the legal framework that supported free trade between Switzerland, the United Kingdom and the European Union, products that used to be unrestrictedly distributed in Europe have become imports having to comply with the relevant legal requirements. Distributors for their part have become importers, and declarations of conformity and CE certificates have lost their validity. Furthermore, notified bodies from Switzerland and the United Kingdom are no longer recognized by the European Commission. Switzerland, the United Kingdom and the European Union have had to grant grace periods to allow regulatory agencies and economic operators to adapt to the new situation.
ConclusionsThe transition period toward the new economic scenario has not yet ended. Both Switzerland and the United Kingdom have had to take stronger measures than the EU to adapt to the changes. Both Switzerland and the United Kingdom are expected to finally incorporate the requirements of the new Regulation in their internal legal systems.
Comparar las medidas que se han tomado por parte de la Unión Europea, Suiza y Reino Unido para mantener la continuidad de mercado cumpliendo con los requisitos regulatorios del Reglamento 745/2017 de Productos Sanitarios.
MétodoPara realizar este trabajo se han revisado las webs oficiales de la Comisión Europea, la Agencia Española del Medicamento y Productos Sanitarios, la Swiss Agency for Therapeutic Products y la Medicines and Healthcare Products Regulatory Agency del Reino Unido y se han realizado búsquedas bibliográficas en PubMed y en internet (Google) con términos como “withdrawal Mutual Recognition Agreement of certificates of conformity European Union Switzerland medical devices, new regulation medical devices UK’ y similares para un periodo comprendido entre enero de 2020 y diciembre de 2021.
ResultadosComo resultado del cese del marco legal que sostenía el libre comercio entre Suiza y Reino Unido de la Unión Europea, la distribución de productos sanitarios se ha convertido en una importación, teniendo que cumplir con los requisitos legales pertinentes. Los distribuidores han pasado a ser importadores, y las declaraciones de conformidad y certificados de Conformidad Europea han perdido su validez. Además, los Organismos Notificados ya no son reconocidos por la Comisión Europea. En consecuencia, Suiza, Reino Unido y la Unión Europea han tenido que conceder periodos de gracia para permitir a las agencias reguladoras y operadores económicos adaptarse a las nuevas condiciones.
ConclusionesEl periodo de transición para la adaptación al nuevo escenario económico todavía no ha concluido. Además, el Reglamento acaba de entrar plenamente en vigor, por lo que se creará normativa de desarrollo que deberá implementarse también en estos países. Por tanto, será necesaria una nueva reglamentación que permita abordar estos aspectos.
Since 2017, the European regulation on medical devices (MD) is Regulation 2017/745 on PS1, hereinafter “Regulation”. In 2002, the Swiss legislation on medical devices (MDs) was brought into line with the European legislation with the enactment of the Medical Device Ordinance (MedDO)2 and the Federal Act on Medicinal Products and Medical Devices3. The equivalence was made possible by the European Union (EU)-Switzerland Mutual Recognition Agreement (MRA) on MD4. The legal basis for this MRA is to be found in the EU directives on MDs5–7. However, the equivalence came to an end on 26 May 2021 with the entry into force of Regulation 2017/745 on MDs1, henceforth the Regulation, which replaced Directive 93/42 on MDs6 and Directive 90/385 on Active Implantable Medical Devices (AIMDs)7.
Consequently, and as a contingency measure, on 19 May the Swiss Federal Council commissioned a thorough overhaul of the MedDO, introduced supplementary provisions, and enacted new rules for the performance of clinical trials with MDs8.
Both the EU and Switzerland have prepared a new MRA on MDs, which has been completed and stands ready to be signed. However, the EU has tied the conclusion of the new MRA to the advancement of the negotiations underway on a new institutional framework agreement9.
As far as in Vitro Diagnostic Medical Devices (IVDMDs) are concerned, no additional legislation is required from Switzerland while the European Directive remains in force5. This will change when the new IVDMD Regulation (2017/746) is enforced on 26 May 202210.
The situation of the United Kingdom (UK), made up of Great Britain (Scotland, England and Wales) and Northern Ireland (NI), is different as, unlike Switzerland, it used to be a member of the EU, which it joined on 1 January 197311.
As a result of the referendum held on 23 June 2016, where UK citizens voted to leave the EU, the British government invoked article 50 of the Lisbon Treaty, informing the European Commission (EC) of its wish to abandon the EU on 29 March 2017. This triggered the so-called Brexit (Britain - exit) process. On 24 December 2020, the EU and the UK concluded a Trade and Cooperation Agreement, aimed at defining the future relations between both parties. This agreement came into force on 1 May 202112. Until then, the UK had followed the European regulations in the realm of MDs, but the new regulations -enacted following the UK transition period- have not been implemented in Britain.
A key point in the EU-UK Withdrawal Agreement was the Northern Ireland Protocol13, designed to avoid the creation of a physical border separating NI from the Republic of Ireland but ensuring at the same time the preservation of the EU Single Market14. By virtue of this Protocol, certain products —including MDs— must comply with the legislation of both the EU and the UK beyond the transition period15.
The purpose of this article was to compare the measures adopted by the EU, Switzerland and the UK to preserve market continuity while complying with the requirements of the Regulation.
MethodsTo carry out the present study, a review was carried out of the official websites of the EC, the Spanish Agency for Medicines and Medical Products (AEMPS), the Swiss Regulatory Agency for Medicines and Health Products (Swissmedic) and the UK's Medicines and Healthcare Products Regulatory Agency (MHRA). A search was also performed in Pubmed for MRAs relative to MDs in Switzerland and the UK, which did not yield any results. Searches were also conducted on the internet (Google), for texts written both in English and in Spanish, using MeSH terms such as “Mutual
Recognition Agreement medical devices Switzerland”, “Mutual Recognition Agreement medical devices UK”, “Northern Ireland Protocol medical devices” during the period extending from January 2021 to December 2021. Texts had to come from the websites of notified bodies (NBs) and regulatory agencies.
ResultsThe information obtained was classified into country-specific sections and under various headings to facilitate comparisons.
European Union - SwitzerlandEver since the MRA4 with that country ceased to apply on 26 May 2021, the EU has regarded Switzerland as a third country in its trade relations with respect to MDs. The Swiss regulatory framework is currently based on a revised MedDO and on the supplementary measures adopted on 19 May 2021 by the Swiss Federal Council together with a new set of rules governing the performance of clinical trials with MDs (CTO MedDO)8. Both modifications came into effect on 26 May 20212. In addition, by virtue of article 82.3 of the Federal Act on Medicinal Products and Medical Devices, the Swiss Federal Council may enforce the EC's measures with respect to the Regulation1 in Switzerland provided that the CE's implementing and delegated acts are directly applicable16.
Requirements for manufacturers and Authorized RepresentativesFrom 26 May 2021, EU manufacturers wishing to introduce or put into service a MD in Switzerland must avail themselves of an importer and appoint a Swiss authorized representative (CH-REP). According to Annex I of the MedDO, aa CH-REP is required for all MDs, including custom-made MDs, kits and systems, as well as for those MDs without a medical purpose.
Economic operators (manufacturers, importers and CH-REPs) must register with Swissmedic within three months from introducing a product into the Swiss market. Once registered, operators are assigned a single registration number (Swiss Single Registration Number, CHRN17, which is the equivalent to the EU's Single Registration Number (SRN). Manufacturers are allowed the following grace periods for appointing a CH-REP9:
- a)
For class III devices, implantable class IIb devices and active implantable devices, until 31 December 2021.
- b)
For non-implantable class IIb devices and for class lla MDs, until 31 March 2022.
- c)
For class I devices, until 31 July 2022.
- d)
For kits and systems, until 31 July 2022.
These grace periods are also applicable to Iceland and Norway, but not to Liechtenstein given the existence of a customs treaty between Liechtenstein and Switzerland18. These periods are not applicable to importers9.
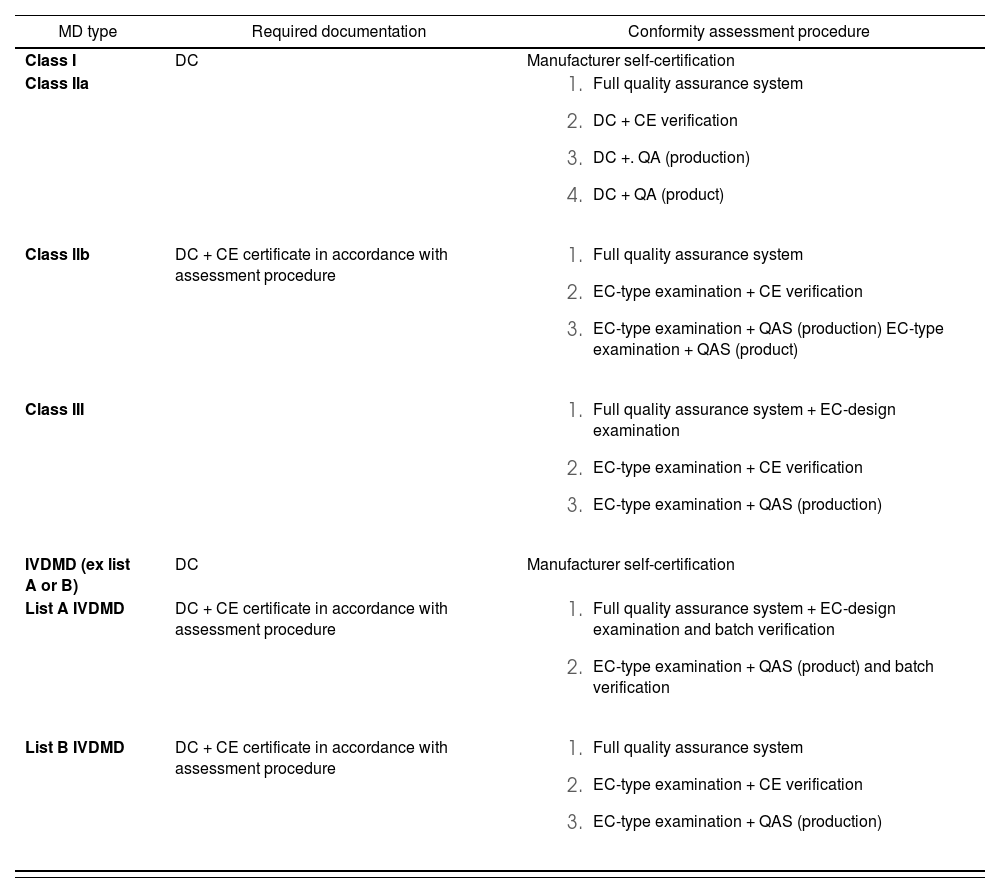
Table 1 summarizes the main differences between the various kinds of certification.
Characteristics of the different medical devices evaluation procedures
| MD type | Required documentation | Conformity assessment procedure |
|---|---|---|
| Class I | DC | Manufacturer self-certification |
| Class Ila |
| |
| Class llb | DC + CE certificate in accordance with assessment procedure |
|
| Class III |
| |
| IVDMD (ex list A or B) | DC | Manufacturer self-certification |
| List A IVDMD | DC + CE certificate in accordance with assessment procedure |
|
| List B IVDMD | DC + CE certificate in accordance with assessment procedure |
|
DC: declaration of conformity; IVDMD: in vitro diagnostic medical device; MD: medical devices; QAS: quality assurance system.
In turn, manufacturers from Switzerland or from third countries who have a CH-REP must appoint an authorized representative in the EU (EC-REP)19. The new data must be reflected in the EU Declaration of Conformity20. Also, from 26 May 2021 distributors will need an import license to carry out these commercial transactions.
Registration of MDsIn Switzerland, manufacturers of custom-made, repacked or relabeled MDs, class I MD, or formerly belonging to class I and reclassified under the new Regulation, have since July 2020 been under the obligation to file a report with Swissmedic21.
In the EU, MDs have so far been registered in the databases of the different member states. An EU-wide database called the European Database on Medical Devices (EUDAMED3)22 is being put together but has not been finalized. This new EU-wide database for MDs will improve the transparency and consistency of the information on MDs. It will comprise six modules covering actor registration, unique device identification (UDI) and device registration, notified bodies and certificates, clinical investigations and performance studies, post-authorization vigilance and market surveillance. The database will store the information required to ensure appropriate market control and an efficient EU-wide surveillance system. At present, all notifications in Spain must be made on the CCPS (Comunicación de Comercialización de Productos Sanitarios) application.
Labeling and conformity markingIn Switzerland, labeling and instructions of use must be printed in the country's three official languages (German, French and Italian)17. The Swiss conformity marking “MD” must appear on the label, although the CE mark is also allowed17,23.
The CH-REP's name must be printed on the outside of the product's package together with the “CH-REP” symbol. Nevertheless, in accordance with an informative note issued by Swissmedic, the CH-REP's name need not appear on the product itself, the user instructions or the other documents accompanying the product24.
In the EU, the Notice to Stakeholders issued by the EC on 26 May 202119 contained a reminder of the obligation to register MDs and of the labeling norms applicable to Swiss MDs. In Spain, the AEMPS issued an informative notice (Nota PS 20/21), where it set the 30th of September of 2021 as the deadline to update all MD labels and user instructions with the new EC-REP or NB20. The deadline has been extended to 30 September 2022 (Note PS 29/21)25.
Notified Bodies and Certificates of ConformityAll certificates issued before 25 May 2017 will remain valid until their expiry or until 26 May 2022 at the latest. Certificates issued from 25 May 2017 under the previous legislation will remain valid until their expiry date or until 26 May 2024 at the latest17.
CE certificates granted to Swiss NBs ceased to be valid on 26 May 202119 as the EU does not recognize them anymore. Declarations of conformity must contain the details of the new EC-REP and certificate number.
Legacy Medical DevicesMDs known as “legacy devices” are those that were introduced into the market in compliance with the existing directives6,7 prior to the entry into force of the Regulation and which can still be legally marketed. This peculiar situation is contemplated in article 120 of the Regulation6,7. Such MDs are:
- –
Class I MDs with a declaration of conformity issued before 26 May 2021 which are required to have a certificate under the new Regulation (e.g., disposable instruments or MDs that have been reclassified by the Regulation).
- –
MDs with a valid CE certificate under the previous regulations.
These legacy products can remain in the Swiss market after 26 May 2021 until their certificate expires, but not beyond 26 May 2024 (they may however be distributed until 26 May 2025). They must comply with the provisions of the MD and the AMID Directives and can only be marketed provided that their design or purpose have not been altered26.
As regards the EU, “legacy devices” are MDs whose manufacturer or authorized representative is Swiss or possesses a certificate of conformity issued by a Swiss NB. According to the EC's Notice to Stakeholders of 26 May 202119, these MDs cannot remain in the European market.
European Union – United KingdomThe UK is made up by Great Britain and NI. The situation of each region will be analyzed separately.
Situation in Great BritainCurrently, the UK regulation in this realm is based on the three old MD Directives5–7, the provisions of which were transposed into UK law as the UK MDR 200227.
- –
Requirements for manufacturers and Authorized Representatives
Manufacturers wishing to sell a MD in the English market must register with the UK's MHRA and appoint a responsible person in the UK (UKRP). Importers and distributors need not appoint a UKRP as they can act in that capacity themselves. When an importer is not the UKRP, he must inform the UKRP about his wish to import MDs and the UKRP must in turn notify who their MD importers are to the MHRA27.
In the EU, UK manufacturers must meet the same requirements as Swiss manufacturers and those with a CH-REP.
- –
Registration of MDs
All MDs, IVDMDs and custom-made MDs must be registered with the MHRA before they can be introduced into the market. MDs already registered with the MHRA need not be registered again (NI manufacturers)27. The MHRA has established certain grace periods for the registration of MDs27:
- a)
AIMDs, class III MDs, implantable class IIb MDs and IVDMDs on list A must be registered from 1 May 2021.
- b)
Non-implantable class IIb MDs class IIa MDs, IVDMDs on list B, and self-diagnosis MDs must be registered from 1 September 2021.
- c)
Class I MDs and general IVDMDs (currently exempted from registration) must be registered from 1 January 2022.
- d)
Manufacturers of class I MDs, custom-made MDs and general IVDMDs which had to be registered prior to 1 January 2021 must still comply with all registration formalities.
UK manufacturers must comply with the same formalities in the EU as Swiss manufacturers or those with a CH-REP.
- –
Labeling and conformity marking
MDs must bear the CE or the UKCA (UK Conformity Assessed) mark, depending on the legislation governing the relevant certificates, and the identifier of the NB or UK Conformity Assessment Body (UKCABs), when applicable27.
Class I MDs and general IVDMDs must be self-certified with the UKCA mark, which will be voluntary until 30 June 2023 and compulsory thereafter27.
MDs may bear both markings (UKCA and CE) even after that date, but the name and address of the UKRP must appear next to the UKCA mark (even in the case of MDs with dual marking)27.
It must be noted, however, that the UKCA marking is not valid in the EU or in NI, where the CE marking (or the UKNI-CE marking in IN) is required27. In Spain, the AEMPS initially gave manufacturers until 30 June 2020 to adapt the labeling and user instructions of their MDs28. Due to implementation difficulties, the deadline was extended until 25 May 202129.
- –
Notified Bodies and Certificates of Conformity
By virtue of UK MDR 2022, the MHRA may appoint an UKCAB for the conduction of conformity assessments for Great Britain, but not assessments related to the CE mark. The exception to this rule is the “UKNI-CE” marking for IN, which is only valid in that region27. The MHRA has automatically converted all the old NBs into UKCABs27.
In addition, the UK government has created a new database, “UK Market Conformity Assessment Bodies”, to replace the EUs NANDO database27.
Regarding the CE certificates issued by an EU-accredited NB, those included in the NANDO database, they will be valid until 30 June 202327. The EU has not recognized UK NBs since 26 May 2021.
Northern IrelandBy virtue of the Northern Ireland Protocol, the requirements to market an MDs in NI are different from those imposed by Great Britain. Due to the particular geographical situation of NI, both the UK and the EU regulations are applicable there.
- –
Requirements for manufacturers and Authorized Representatives
Manufacturers and authorized representatives based in the EU/EEA must appoint a single UKRP for dealing with MDs to be marketed in NI. Appointing a UKRP is not required if the manufacturer is based in Great Britain or NI, or if they have a Northern Ireland-based Authorised Representative (NIAR). Nor is it necessary if they market general class I, custom-made or general IVDMDs that are registered with a competent EU authority.
Similarly, manufacturers from Great Britain wishing to introduce a MD in the NI market must appoint an EC-REP or a NIAR15. A single entity may act as both a NIAR and a UKRP.
In cases where a NI importer has appointed a NIAR or a UKRP, the importer must inform them of his intention to import the relevant MDs. The NIAR or UKRP must in turn provide the MHRA with a list of the importers they work for15.
NI-based manufacturers do not require an EC-REP to market their products in the EU. In cases where the MD requires to be assessed by a NB, the latter must be EU-based15. UK or third-country manufacturers may appoint an EC-REP in NI for the EU.
- –
Registration of MDs
Most general custom-made or general IVDMD marketed in NI must be registered with the MHRA. This will depend on where the manufacturer or the authorized representative is based, and on the class of MDs to be marketed. The same grace periods apply in NI as in the UK15.
If an NI-based authorized representative is appointed, the manufacturer must register all MD classes with the MHRA. However, if an EU-based authorized representative is appointed, all MD classes must be registered other than class I, custom-made or general IVDMD.
The EU does not impose additional requirements to those in the Regulation.
- –
Labeling and conformity marking
The UKCA marking is not valid in NI. In NI, all MDs requiring to be assessed by a third party must bear the UKNI mark in addition to the CE mark if the conformity assessment was performed by a UKCAB27. The UKNI mark is not valid on its own. If the assessment is performed by an EU-accredited NB, the MD will only bear the CE mark. This means that the UKNI mark will only accompany an MD if the product will be marketed in NI, it requires a conformity assessment and the latter was carried out by a UKCAB27.
The EU does not accept the UKNI marking, MDs intended for the EU market are required to bear only the CE mark27.
- –
Notified Nodies and Certificates of Conformity
The UK's notification bodies may carry out conformity assessments only for the NI market, not for the EU market. To be valid, a CE mark on an MD intended for the NI or the EU market must have been assessed by an EUapproved NB.
The UK government has guaranteed that all NI MDs can be marketed in the UK. This means that MDs bearing NI's conformity mark can be legally marketed in the whole of the UK territory even beyond 30 June 2023.
According to the EU regulation, Irish NBs must issue their certifications in compliance with the Regulation.
Table 2 summarizes the most important changes regarding the marketing of MDs in the EU, Switzerland and the UK after the entry into force of the new Regulation.
Main changes and measures adopted by the European Union, Switzerland and the United Kingdom
| Current legislation | Conformity marking | Authorized representative | Certificates | Labeling | |
|---|---|---|---|---|---|
| Switzerland |
|
|
|
|
|
| Great Britain | MD regulation of 2002 (UK MDR 2002) |
| A UKRP is required for manufacturers from outside Great Britain but not for importers or distributors. These are allowed to act as UKRPs |
|
|
| Northern Ireland |
| − CE mark is compulsory In addition, a UKNI mark is required for MDs certified by a UKCAB The UKCA is not recognized MDs with a UKNI mark are valid throughout the UK territory beyond 30 June 2023 |
| CE certificates remain valid |
|
CH-REP: Authorized Representative in Switzerland; EC-REP: Authorized Representative in the European Union; EEA: European Economic Area; EU: European Union; MD: medical device; MedDO: Medical Device Ordinance; NB: notified body; NI: Northern Ireland; UK MDR 2002: UK Medical Device Regulation 2002; UK: United Kingdom; UKRP: UK Responsible Person.
The end of free MD trade between the EU and Switzerland and the UK has had a negative impact on both economic operators and patients. This article provides a comparison of the measured adopted at this time of great confusion, to comply with the newly introduced requirements governing the marketing of MDs in the EU, Switzerland and the UK after the entry into force of the Regulation.
The two main consequences of Switzerland and the UK becoming third countries are, on the one hand, the risk of supply shortages and, on the other, the need to create economic operator and MD registries as well as other infrastructures such as databases that allow these countries to control their internal market.
Given its size, the EU is at an advantage over Switzerland and the UK. EU authorities have immediately and unilaterally terminated the validity of CE certificates issued by NBs based in these countries, making it illegal to market Swiss and UK MDs. However, the opposite has not been possible.
Simultaneous loss of validity of CE certificates entails a significant additional workload for EU NBs and a delay in the issuance of new certificates. Companies that did not act in anticipation experienced a cessation of their trade relations until they managed to obtain the corresponding certificates and to update the technical documentation.
At the same time, according to Sidley Austin's memorandum titled EU/ EEA Market Access for “Swiss Legacy” Devices post-abandonment of Swiss EU MRA30“the Commission's unilateral decision to cease application of the MRA, and the Commission's purported retrospective withdrawal of mutual recognition for Swiss Legacy Devices, as expressed in particular in the Notice to Stakeholders, are contrary to EU law, the MRA and WTO law”30. Although this is a valid argument, it is inly applicable to legacy devices with CE certificates from non-Swiss NBs. To our knowledge, the EU has made no statement about these claims.
On the other hand, there is the need for Switzerland and the UK to create economic operator and MD registers that allow them appropriate control and surveillance of their internal market. Creation of these databases should be a priority for these countries, as should be the holding of negotiations conducive to gaining some measure of access to EUDAMED3, as both Switzerland and the UK are part of the European continent and any incident occurring in Europe could have direct consequences for the two countries.
It is to be expected that both Switzerland and the UK will adapt to the regulations by means of their internal legislation as any other decision would mean imposing additional trade barriers, which would have a negative impact on their patients’ health.
The importance of the present study is that it analyses in one single document the main regulatory changes introduced by the EU, Switzerland and the UK and the different transition periods they have granted to economic operators and regulatory agencies as a result of the regulatory divorce that occurred in the realm of MDs between the EU, Switzerland and the UK on 26 May 2021.
In short, although the reasons why UK and Switzerland decided to separate from the EU are different, both countries have been forced to adapt to the provisions of the Regulation to avoid more than likely supply shortages.
The EU has asserted its strength by immediately cancelling all CE certificates issued by Switzerland and the UK and demanding new certifications by EU-accredited NBs, albeit granting them a grace period. In the case of Switzerland, doubts exist regarding the lawfulness of this measure.
In turn, both Switzerland and the UK have been forced to establish longer transition periods to give regulatory agencies and economic operators enough time to adapt their MDs to the new national legislations. In practice, this means that these countries still accept MDs bearing a CE mark and certificates issued by EU-based NBs; acceptance will be permanent in Switzerland and will be terminate on 30 June 2023 in the UK.
As implementation of the EU Regulation progresses, these two countries are likely to adopt additional contingency measures. Further studies will be required to evaluate the impact of such measures.
FundingWithout funding.
AcknowledgementsWe would like to thank Nadia Chahboune for her participation in the review of the article, contributing her knowledge and professional experience.
Conflict of interestsThe authors declare that there is no conflict of interest with other persons or entities.
Contribution to the scientific literature
This study is intended to shed light on the new regulatory framework governing trade relations between the European Union, Switzerland and the United Kingdom with respect to medical devices. The coming to an end of the Mutual Recognition Agreement with Switzerland has occurred simultaneously with the entry into force of new Regulation 2017/745 on medical devices. Moreover, since January 2021, the UK has not been a member of the European Union. Economic operators must adapt to the new requirements of the Regulation and of each individual country, even if such requirements are not clearly specified in any official document.
Early Access date (06/25/2022).

