


The aim of the study was to evaluate the safety profile of nirmatrelvir-ritonavir (NMV-r) in real clinical practice and to analyse the clinical relevance of drug–drug interactions in the development of adverse events.
MethodsObservational, retrospective study in which safety data of patients treated with NMV-r between April and July 2022 in an outpatient setting were evaluated. The duration of follow-up was 28 days and the number of adverse reactions reported, as well as whether they were managed on an outpatient basis or required health care, and the presence of renal and hepatic function impairment were assessed. Concomitant treatment was reviewed, identifying theoretical drug–drug interactions (TDDIs) whose severity was defined using the Lexi-interact classification.
ResultsThe study included 146 patients. 82 (56.16%) were women, whose median age was 65 years (22–95). the number of TDDIs detected and maintained during treatment with NMV-r was 164, with the percentage of patients with at least 1 interaction being 62.33%. The median number of TDDIs per patient was 1 (0–5). At least 1 adverse event (AE) was reported in 18 patients (11.84%). 11 AEs were potentially related to any TDDI. 7 patients required contact with hospital assistance for AE management. 8 patients had impaired renal function and 2 had impaired liver function at 28 days. The main groups of drugs implicated in the occurrence of an AE were oral anticoagulants and calcium antagonists.
ConclusionsOur results show a high number of TDDIs detected were detected between NMV-r and other drugs. This study provides greater knowledge of the drugs involved in such interactions and their potential relationship with the occurrence of adverse events.
evaluar el perfil de seguridad de nirmatrelvir-ritonavir (NMV-r) en la práctica clínica real y analizar la relevancia clínica de las interacciones farmacológicas en el desarrollo de eventos adversos.
Material y métodosestudio observacional, retrospectivo en el que se evaluaron los datos de seguridad de pacientes tratados con NMV-r entre abril y julio de 2022. Se recopilaron datos demográficos y analíticos antes de comenzar el tratamiento. La duración del seguimiento fue de 28 días y se evaluó el número reacciones adversas reportadas, así como si fueron manejadas de forma ambulatoria o precisaron de asistencia sanitaria especializada, y la presencia de deterioro de la función renal y hepática. Se revisó el tratamiento concomitante, identificando interacciones farmacológicas teóricas (IFT) cuya gravedad fue definida mediante la clasificación Lexi-interact.
ResultadosEl estudio incluyó 146 pacientes. 82 (56,16%) eran mujeres, cuya mediana de edad fue de 65 años (22–95). El número de IFT detectadas y mantenidas durante el tratamiento con NMV-r fue de 164, siendo el porcentaje de pacientes con al menos una interacción de 62,33%. La mediana de IFT por paciente fue de 1 (0–5). En 18 pacientes (11,84%) se reportó al menos un evento adverso (EA). 11 EA se relacionaron potencialmente con alguna IFT. 7 pacientes requirieron contacto con asistencia hospitalaria para el manejo del EA. 8 pacientes presentaron deterioro de la función renal y 2 de la función hepática a los 28 días. Los principales grupos de fármacos implicados en la aparición de algún EA fueron los anticoagulantes orales, así como los calcio-antagonistas.
Conclusionesnuestros resultados muestran un elevado número de IFT detectadas entre NMV-r y otros fármacos, aunque la frecuencia de eventos adversos asociados fue baja. Este estudio proporciona un mayor conocimiento de los fármacos implicados en dichas interacciones y su potencial relación con la aparición de eventos adversos.
Nirmatrelvir/ritonavir (NMV-r) is an antiviral medication approved by the European Medicines Agency (EMA) for adults who do not require supplemental oxygen and who are at high risk of progression to severe coronavirus disease 2019 (COVID-19), an infectious disease that has caused more than 6 million deaths worldwide.1 According to the results of the EPIC-HR clinical trial—a Phase 2/3 placebo-controlled study—NMV-r has demonstrated efficacy: treatment with this medication showed an 89% relative reduction in the risk of hospitalisation or death from any cause within 28 days of starting treatment.2 This trial found that there were numerically more suspected adverse events (AEs) related to NMV-r than to placebo. Currently, NMV-r is the treatment of choice in patients who are immunosuppressed or at high risk of progressing to severe forms of COVID-19. The standard dose is 300 mg nirmatrelvir+100 mg ritonavir q12h for 5 days. Nirmatrelvir acts by blocking SARS-CoV-2 pseudo-3C protease and is metabolised in humans via the cytochrome P450 (CYP3A4) enzyme pathway.3 Ritonavir is a known potent inhibitor of this enzyme and is therefore used in the treatment of HIV infection to enhance the effect of protease inhibitors; however, it is well known for its ability to generate potentially serious interactions with many other drugs.4 In fact, ritonavir is involved in drug–drug interactions (DDIs) in approximately 30% of the general population exposed to this drug, rising to over 60% in individuals of more than 60 years of age.5 There are various algorithms for the management of drug interactions in patients treated with NMV-r6,7; however, despite theoretical knowledge of such interactions, there are currently few studies that have analysed their clinical relevance in routine practice. In addition, the recent use of ritonavir in SARS-CoV-2 infection means that the drug may be prescribed by physicians unfamiliar with its use, leading to potential AEs due to inadvertent DDIs at the time of prescribing treatment. This study evaluated the safety profile of NMV-r in real-life clinical practice, collected information on the theoretical DDIs detected and AEs observed, and analysed the clinical relevance of DDIs in the development of AEs.
MethodsObservational retrospective study in which safety data were evaluated in a cohort of 146 patients treated consecutively with NMV-r between April and July 2022. Treatment with NMV-r was administered at a dose of 300 mg nirmatrelvir+100 mg ritonavir q12h for 5 days; however, in patients requiring dose adjustment for impaired renal function, the dose was 150 mg nirmatrelvir+100 mg ritonavir q12h for 5 days. The study was conducted in outpatients treated in a class 58 reference hospital with 1300 beds and an assigned population of 450 000. The following data were collected: demographic data (age and sex); analytical data (estimated glomerular filtration rate and transaminases) before starting treatment; prioritisation level according to the criteria published by the Spanish Agency of Medicines and Health Products (AEMPS); and the hospital protocol: patients who do not require supplemental oxygen; patients presenting within the first 5 days of symptoms; immunocompromised patients; or patients at risk of progressing to severe COVID-19. The follow-up period was 28 days. During this period, we assessed the number of adverse events reported, the need for both primary and specialised healthcare, and renal and liver function. Adverse events were classified according to the Common Terminology Criteria for Adverse Events of the US National Cancer Institute (version 5.0). Concomitant drug treatment was reviewed for all patients included in the study. Theoretical DDIs (i.e. the study topic) attributed to drugs that were not discontinued were quantified and classified according to the World Health Organisation (WHO) Anatomical Therapeutic Chemical (ATC) code. The severity of each interaction was assessed using the Lexi-interact module classification provided by the open-access, international Lexicomp database, which includes information on the level of urgency and actions to be taken in the event of an interaction.9 In the pharmaceutical validation process, the module recommends discontinuing treatment with drugs that may interact with each other in a clinically significant manner and where the risk/benefit ratio could be unfavourable (Lexi-Interact risk-rating: D/X). The study data were collected from the prescribing software and hospital's medical records. The statistical analysis was descriptive. Qualitative variables are expressed as absolute and relative frequencies and quantitative variables are expressed as medians and interquartile ranges. The study was approved by the hospital's Drug Research Ethics Committee (reference 23/150), thus foregoing the need for written informed consent and allowing the use of previously collected and anonymised data. The results are reported according to STROBE (Strengthening the Reporting of Observational Studies in Epidemiology) guidelines.
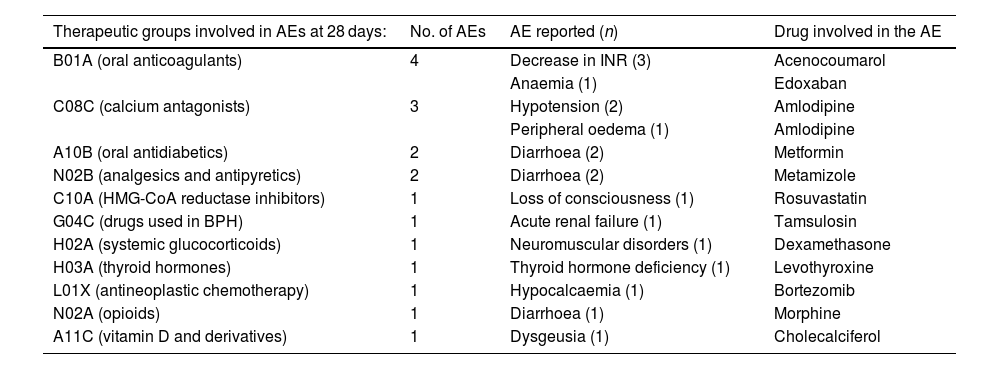
ResultsOf the 146 patients included in this study, 82 (56.1%) were women (median age: 65 [22–95] years). In total, 27 (18.69%) patients with creatinine clearance values ranging from 30 mL/min to 60 mL/min required pre-treatment dose adjustment. (150 mg nirmatrelvir+100 mg ritonavir q12h for 5 days). In total, 106 (72.60%) patients met the indication criteria because they were in AEMPS10 Group 1, which prioritises adults with high-risk conditions. This group includes immunocompromised patients irrespective of their vaccination status. Thirty patients (20.54%) belonged to Group 4, which included patients of more than 80 years of age with at least 1 risk factor for progression to severe forms. One patient (0.68%) was not vaccinated. In total, 164 theoretical DDIs were detected during treatment with NMV-r and treatment was continued; 62.33% of patients had at least 1 interaction. The median number of interactions per patient was 1 (0–5). Among the theoretical DDIs that did not lead to drug discontinuation, 140 (85.37%) were class C (monitor therapy), 18 (10.98%) class D (consider modifying therapy), and 6 (3.66%) class X (contraindicated); in the latter case, a joint assessment by the physician and pharmacist was that treatment should be continued in view of the favourable risk–benefit ratio. Table 1 shows the AEs detected, their relationship with interactions, the need for healthcare, and the number of patients with impaired renal and/or hepatic function. Table 2 shows the pharmacotherapeutic groups that potentially produced AEs, the reported AE, and the drug involved.
Overall summary of adverse events.
| Variable | n = 146 (%) |
|---|---|
| Patients with adverse events at 28 d | 18 (11.84) |
| Adverse events potentially related to interactions | 11 (61.1) |
| Patients with outpatient management of adverse events | 11 (7.53) |
| Patients requiring contact with healthcare (primary care or hospital) | 7 (4.60) |
| Patients with deterioration of renal function at 28 d | 8 (7.02) |
| Patients with deterioration of liver function at 28 d | 2 (1.75) |
| Patients with discontinuation of nirmatrelvir-ritonavir due to adverse events | 2 (1.37) |
Reported adverse events involved in theoretical DDIs by therapeutic group.
| Therapeutic groups involved in AEs at 28 days: | No. of AEs | AE reported (n) | Drug involved in the AE |
|---|---|---|---|
| B01A (oral anticoagulants) | 4 | Decrease in INR (3) | Acenocoumarol |
| Anaemia (1) | Edoxaban | ||
| C08C (calcium antagonists) | 3 | Hypotension (2) | Amlodipine |
| Peripheral oedema (1) | Amlodipine | ||
| A10B (oral antidiabetics) | 2 | Diarrhoea (2) | Metformin |
| N02B (analgesics and antipyretics) | 2 | Diarrhoea (2) | Metamizole |
| C10A (HMG-CoA reductase inhibitors) | 1 | Loss of consciousness (1) | Rosuvastatin |
| G04C (drugs used in BPH) | 1 | Acute renal failure (1) | Tamsulosin |
| H02A (systemic glucocorticoids) | 1 | Neuromuscular disorders (1) | Dexamethasone |
| H03A (thyroid hormones) | 1 | Thyroid hormone deficiency (1) | Levothyroxine |
| L01X (antineoplastic chemotherapy) | 1 | Hypocalcaemia (1) | Bortezomib |
| N02A (opioids) | 1 | Diarrhoea (1) | Morphine |
| A11C (vitamin D and derivatives) | 1 | Dysgeusia (1) | Cholecalciferol |
AE, adverse event; INR, International Normalised Ratio; BPH, benign prostatic hyperplasia, DDI, drug–drug interaction.
Of the 146 patients, 76 (52.05%) did not experience any AE even though they remained on a drug that potentially interacted with NMV-r. Fig. 1 shows the number of detected DDIs that did not result in AEs in the study patients.

Grade 3 or 4 AEs were detected in 3 patients: NMV-r and tamsulosin interaction in 1 patient caused acute renal failure requiring hospital admission; treatment with amlodipine in another led to hypotension, which was managed in the primary care setting; and a patient treated with bortezomib developed grade 3 hypocalcaemia requiring an emergency department visit and treatment with intravenous calcium gluconate.
DiscussionThere was a lower incidence of any AE in our study than in the pivotal trial (12.33% vs 22.6%, respectively) and fewer grade 3 or 4 AEs (1.3% vs 4.1%, respectively).2 Our results may be attributed to the lack of proactive follow-up, as we relied on patient surveys, which differs from the follow-up conditions employed in the clinical trial. The main differences between our patients and those enrolled in the EPIC-HR trial2 were that our patients had a higher median age (65 years vs 44 years) and almost all of them had been vaccinated (99% vs 0%), without correlation with the variables analysed in our study.
A recent review summarised the likelihood of having a clinically relevant DDI according to the drugs involved.11 The results of our study are consistent with those of this review, as it referred to the low potential for clinically significant DDIs between therapeutic groups such as oral antidiabetics and glucocorticoids. In our cohort, both therapeutic groups were found to have the highest rate of potential DDIs without harm. On the other hand, the review established the potential for relevant DDIs between NMV-r and antihypertensives such as calcium antagonists, whose AUC can increase by up to 2-fold: and in our population, an AE occurred in 21% of patients treated with calcium antagonists, although 11 such patients did not experience any AE.
This review also addressed oral anticoagulants and antiplatelet agents: in our study, these were the main groups involved in clinically relevant DDIs. Specifically, 3 out of 4 patients treated with NMV-r and acenocoumarol experienced a decrease in their INR, necessitating an adjustment in their antivitamin K dose. This result is in line with those of previous studies,12 in which the INR was measured in 29 patients treated with NMV-r and warfarin. The median INR before and after NMV-r administration was 2.4 (1.1–3.7) and 1.95 (1.5–2.3), respectively: approximately half of the patients experienced sub-therapeutic INR values (target INR 2–3).
Given the high percentage of patients in whom DDIs were detected, we recommend that their treatment with NMV-r should be carefully managed, given that these patients are typically elderly and polymedicated. Such management typically involves dose reductions, the discontinuation of some drugs, or increased monitoring of adverse events. In the study population, many drugs were discontinued, and their reintroduction was recommended 3 days after the end of NMV-r treatment.
In our study, there was no correlation between the high number of DDIs detected and the low incidence of AEs related to these DDIs. Although the lack of proactive follow-up may have contributed to this result, it is more likely due to the low dose of ritonavir used (100 mg) compared to that used in HIV treatment (200–400 mg/d), as well as to the short treatment regimen of 5 days, which contrasts with the regimen used in chronic HIV treatment. It has been observed that ritonavir reaches its peak inhibitory effect on CYP3A4 at 48 h,13 whereas its effect on inducing CYP2C9 or CYP2C19 enzyme activity reaches its peak between 5 and 7 days of treatment. The resulting DDIs that occur through induction may be less clinically relevant.
One of the main limitations of our study is its retrospective nature. Nevertheless, some of the AEs in patients with theoretical DDIs may not have been exclusively related to such interactions, given that some AEs form part of the intrinsic safety profile of these drugs, such as diarrhoea in the patients receiving metformin and dysgeusia in the patient receiving cholecalciferol. In order to maintain homogeneity in data analysis, a single database was used to classify interactions according to severity. The main strength of this study is that it provides a better understanding of the drugs involved, the nature of the DDIs, and their potential effects. These aspects may help to design strategies for the early detection of higher risk groups of patients on medication, improve outcomes, and support clinical decisions based on real-life experience.
ConclusionsThe results indicate a substantial number of theoretical DDIs between NMV-r and other drugs, although there were few associated AEs. This study provides a better understanding of the drugs involved in such interactions and their potential relationship with AEs. Hospital pharmacists could play a key role in the pharmacotherapeutic management of patients treated with NMV-r and in the prevention of AEs; nevertheless, future studies are needed to assess the effect of pharmacist review of DDIs on health outcomes.
Although the urgent need to respond to the health crisis posed by COVID-19 resulted in a surge in scientific research, further studies are currently needed on its pharmacological management, in which hospital pharmacy services could play a key role.14,15
Contribution to the scientific literatureThis study describes real-life experience with nirmatrelvir-ritonavir. This combination medication is used in the treatment of patients at high risk of developing severe SARS-CoV-2 infection. It has received conditional approval by the EMA; however, the management of potential DDIs and their role in the development of AEs remain unclear. The reported evidence of adverse events, combined with the analysis of potential DDIs identified in this article, may help optimise drug utilisation and guide clinical decision-making in the management of COVID-19 patients.
Author statementÁlvaro González Gómez and Jose Manuel Caro-Teller contributed to the original concept of the study, its design, data collection, analysis, interpretation, writing, revision, and data approval.
Iván González Barrios contributed to the design of the study, data collection and analysis, writing, and critical review of the article.
Almudena Castro Frontiñán contributed to data interpretation, writing, revision, and critical review of the article.
Pedro Pablo Rodríguez Quesada contributed to reviewing the article.
José Miguel Ferrari-Piquero contributed to data analysis and interpretation, writing, and critical review of the article.
All authors approved the final version of the article.
FundingNone declared.
Responsibilities and assignment of rightsAll authors accept responsibility as defined by the International Committee of Medical Journal Editors (available at http://www.icmje.org/).
In the event of publication, the authors grant exclusive rights of reproduction, distribution, translation, and public communication (by any audio, audiovisual, or electronic means or support) of their work to Farmacia Hospitalaria and, by extension, to the SEFH.
Ethical responsibilitiesThe study was approved by the Hospital's Drug Research Ethics Committee (ref. 23/150), thus foregoing the need for written informed consent and allowing the use of previously collected and anonymised data.

