



La hemofilia A adquirida (AHA), a diferencia de la hemofilia A (HA), es un trastorno hemostático de origen autoinmune que se da tanto en hombres como en mujeres. Es una enfermedad rara, poco conocida, infradiagnosticada y con un alto índice de mortalidad1. Se origina cuando el organismo desarrolla anticuerpos frente al factor VIII (inhibidor FVIII). En el 52% de los casos se desconoce la causa. En el resto, se ha relacionado con enfermedades autoinmunes, cáncer, embarazo, vacunas y/o con tratamientos farmacológicos2. Las manifestaciones son variadas, desde sangrado intenso (generalmente tras traumatismos o el parto), hasta sangrados espontáneos de localización y gravedad variadas. El diagnóstico es difícil, por desconocimiento y porque a menudo queda enmascarado, como en los pacientes anticoagulados3. Un diagnóstico rápido es fundamental para el inicio precoz de la terapia antihemorrágica4.
Los pilares del tratamiento son agentes hemostáticos diferentes al FVIII, para restablecer la hemostasia, y la terapia inmunosupresora, para erradicar los inhibidores5. La terapia hemostática en los pacientes que presentan hemorragias graves o la necesidad de una cirugía urgente se basa en agentes bypass (complejo de protrombina activado o rFVIIa) o como alternativa, el FVIII recombinante porcino, con menor riesgo trombótico. El tratamiento inmunosupresor para erradicar el inhibidor consiste en corticoides en monoterapia o asociados a ciclofosfamida y/o rituximab. El tratamiento combinado se asocia a una respuesta más rápida, pero con mayor riesgo de complicaciones5.
Emicizumab, anticuerpo monoclonal biespecífico que mimetiza la actividad del FVIII, no tiene indicación en AHA; sin embargo, por extrapolación de los resultados en HA con inhibidor, puede ser una opción válida en esta enfermedad.
Descripción del casoSe presenta el caso de un paciente varón de 66 años, que acude a urgencias derivado de atención primaria por edemas en los miembros inferiores y mal estado general, con antecedentes de cirrosis hepática por enolismo. Ingresa en la unidad de digestivo con diagnóstico de esofagitis péptica severa y hernia de hiato gigante. Durante los primeros días del ingreso se objetiva un hematoma espontáneo extenso en la cara posterior del muslo izquierdo, con edema y molesto en la palpación. El paciente no había presentado alteraciones hemostáticas ni anemización en los días previos. A las 72 h mejora de forma espontánea, pero luego aparece otro hematoma submandibular acompañado de odinofagia que evoluciona a sangrado leve en la boca. La exploración por cirugía maxilofacial revela entonces un hematoma subcutáneo submental y en el suelo de la boca, sin aparente colección, tumefacción, induración ni signos de infección. En ese momento, el equipo clínico decide no modificar el tratamiento.
Durante 17 días el paciente presenta nuevos hematomas espontáneos que van aumentando de tamaño en distintas localizaciones (el cuello, el muslo, el brazo y el costado), sin traumatismos que lo justifiquen. Analíticamente, presenta anemia con hemoglobina de 6,5 g/dl, por lo que se administran 10 dosis de hierro intravenoso (200 mg cada 48 h) y 10 transfusiones de sangre.
Tras ser evaluado por el equipo de hematología, se diagnostica de AHA. El paciente no presentaba antecedentes oncológicos, autoinmunes u otros factores de riesgo asociados a AHA, por lo que se considera un caso de AHA idiopática. Del estudio básico de coagulación en el diagnóstico sobresalen: tiempo de tromboplastina parcial activada 80 s, tiempo de protrombina 13,2 s y fibrinógeno 292 mg/dL. De las pruebas de coagulación especial destacan: anticoagulante lúpico negativo, actividad FVIII 9% y unidades Bethesda 1,901. Se decide iniciar el tratamiento inmunosupresor con metilprednisolona intravenosa (2 dosis de 90 mg cada 12 h como dosis de carga, para continuar con 80 mg diarios vía oral) y soporte hasta control hemostático con agente bypass (rFVIIa, 5 mg cada 6 h por vía intravenosa) (fig. 1). Ante un empeoramiento del hematoma costal, se opta por administrar 2 concentrados de hematíes.
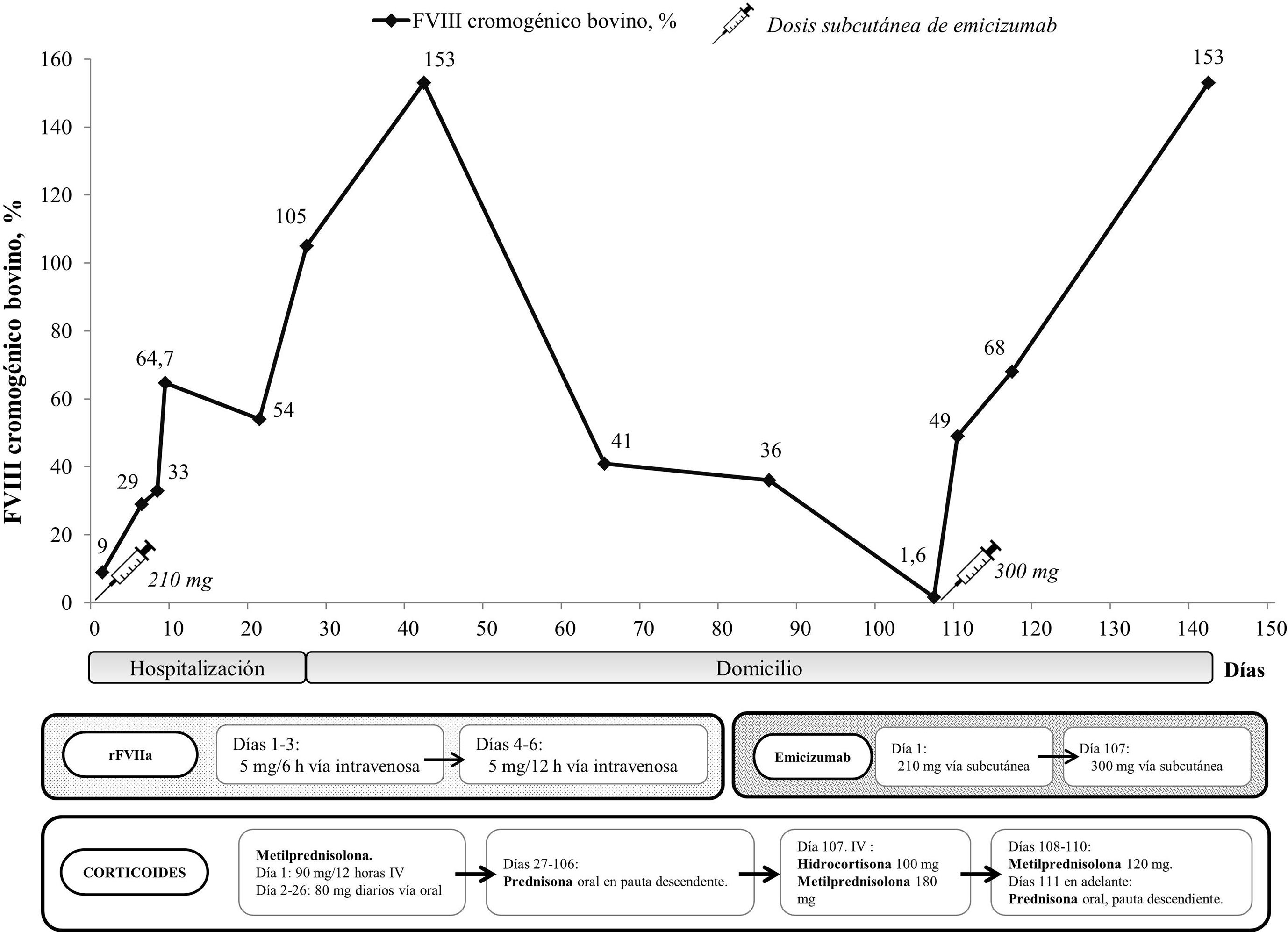
Se decide entonces iniciar el tratamiento con emicizumab, como uso fuera de indicación. Tras obtener el consentimiento informado del paciente, se administra una dosis de 210 mg de emicizumab (3 mg/kg) por vía subcutánea6. El tratamiento con rFVIIa se mantiene 3 días cada 6 h y otros 3 días cada 12 h. La actividad de FVIII se determina mediante el método cromogénico bovino, ya que no interfiere con emicizumab7.
Ante la mejoría paulatina de los hematomas, el buen rendimiento hemostático desde el inicio del tratamiento (actividad FVIII: 105%) y la ausencia de nuevos sangrados, las unidades de hematología y cirugía consensuan el alta hospitalaria tras 2 meses de ingreso, 27 días después de la administración de emicizumab. Al alta, se indica un tratamiento con 40 mg diarios de prednisona oral.
En la primera consulta de seguimiento a los 15 días, el paciente no muestra sintomatología de sangrado (ni con el cepillado dental, ni en la orina o las heces), ni hematomas espontáneos, con actividad de FVIII 153,3% y nivel de emicizumab 25,6 μg/ml (42 días postadministración), por lo que se reduce la dosis de prednisona a 20 mg diarios. En las revisiones periódicas quincenales, se va observando una mejoría general sin sangrados.
En una visita de seguimiento a los 2 meses del alta, se detecta una recaída relacionada con problemas de adherencia al tratamiento corticoide, al objetivarse un gran hematoma sublingual de varios días de evolución con odinofagia (actividad de FVIII: 1,6%). Se administra de forma urgente una dosis intravenosa de hidrocortisona de 100 mg y una segunda dosis de emicizumab (300 mg = 4,3 mg/kg). Posteriormente se administra una dosis de carga de 180 mg de metilprednisolona intravenosa, para continuar en el domicilio con 120 mg diarios vía oral. El paciente presenta evolución favorable en la siguiente cita (a las 72 h) y en las sucesivas, por lo que se reduce la pauta de corticoides.
A los 4 meses del alta, no se vuelven a producir incidencias relacionadas con la hemostasia, con una actividad de FVIII >100%, por lo que se suspende el tratamiento inmunosupresor y se programan controles con frecuencia mensual.
DiscusiónEl manejo de la AHA continúa siendo un reto desde el punto de vista de la seguridad y de la eficacia, precisando de agentes más seguros y con una posología que reduzca la dependencia de la terapia hemostática endovenosa y posibilite un control ambulatorio, tal y como ha sucedido con el tratamiento de la HA, lo cual ha supuesto un cambio de paradigma en la enfermedad.
El papel potencial de emicizumab en AHA no ha sido estudiado, ni está autorizado en la ficha técnica. Los pocos casos publicados (ninguno en España) sugieren que podría ser una opción terapéutica segura y eficaz, consiguiendo reducciones significativas en los índices de sangrado8,9. Una de las principales limitaciones para su uso radica en la elección de las dosis y la frecuencia de administración, ya que aún no se dispone de recomendaciones para el uso del fármaco en esta indicación6. Por otro lado, el riesgo de trombosis no ha sido evaluado en estos pacientes, aunque en los ensayos pivotales que condujeron a la comercialización del fármaco, los casos de complicaciones trombóticas se asociaron al uso concomitante del complejo de protrombina activado, no del rFVIIa. No obstante, hay un ensayo clínico en fase de reclutamiento y otro completado, con resultados pendientes de publicar (NCT05345197, NCT04188639), que sin duda arrojarán luz sobre el uso en AHA.
En el caso que nos ocupa, se realizó inmunosupresión con corticoides en monoterapia y como terapia hemostática se empleó rFVIIa durante 6 días y se administraron 2 dosis de emicizumab con un intervalo de 105 días entre ambas10. El uso de emicizumab resultó seguro, posiblemente contribuyó a acortar la estancia hospitalaria y permitió un tratamiento inmunosupresor menos agresivo, con menos complicaciones.
En conclusión, el empleo fuera de indicación de emicizumab en AHA parece presentar un perfil de eficacia y seguridad favorable para su futuro uso como tratamiento estándar. Se necesitan ensayos clínicos y seguimiento a largo plazo para evaluar el riesgo-beneficio y establecer las pautas de tratamiento.
FinanciaciónSin financiación.
AutoríaLos autores firmantes han contribuido a la elaboración del artículo en cuanto a la concepción y diseño del caso clínico descrito. M. Ángeles Ocaña Gómez y Jorge Esquivel Negrín se han encargado de la escritura del artículo. Asimismo, Mario Ríos De Paz y M. Dolores De Dios García han realizado una revisión crítica del artículo con importantes contribuciones intelectuales. Los 4 autores firmantes han aprobado la versión final de cara a su publicación. La recogida de datos se efectuó por M. Ángeles Ocaña Gómez, Jorge Esquivel Negrín, Mario Ríos De Paz y M. Dolores De Dios García.
Conflicto de interesesLos autores declaran no poseer conflictos de intereses.
Presentaciones en congresos/reuniones científicasNo.
Aportación a la literatura científicaInformar sobre un caso clínico de hemofilia A adquirida que ha sido tratado con emicizumab. Exponer dosis utilizadas y resultados obtenidos.
La indicación autorizada de emicizumab en hemofilia A con inhibidor es la referencia para su uso, por lo que todos los resultados clínicos que se publiquen en otras indicaciones contribuirán a diseñar la mejor estrategia de tratamiento.

