
To conduct an assessment of cyclodextrins used as excipients in pharmaceutical formulations, considering compounding, biopharmaceuticalpharmacokinetic, toxicological, regulatory, economic and commercial aspects.
MethodsA literature search was performed of highly cited review and original articles. Regulatory and legislative documents and well-established pharmacopoeias were also consulted.
ResultsSolubility, resistance to hydrolysis and complexation efficiency are variables that depend on the cyclodextrin itself and on the drug to be complexed. In some cases, addition of excipients is necessary to improve complexation efficiency. Complexation of drugs with cyclodextrins at laboratory scale tends to be rather inconsistent. Moreover, wide variations exist for the same cyclodextrin across different suppliers and even across batches from the same supplier. This means more control analyses must be carried out of pharmaceutical preparations. Problems with the stability of cyclodextrin-drug complexes could affect the oral bioavailability of the drugs. Additionally, some cyclodextrins may optimize the drug's permeability through specific biological membranes and the length of time it remains in contact with them. Despite the safety profile of cyclodextrins, exceeding certain dosing thresholds and administration times might cause adverse effects. Only cyclodextrins recognized as excipients by well-established pharmacopeias should be used in pharmaceutical compounding. Cyclodextrins lead to an increase in the global costs of compounding and their purchase through recognized suppliers is often unfeasible.
ConclusionsDespite their interesting properties as excipients due to inclusion complex formation, the need to carry out more quality control analyses and stability constant studies, combined with the high cost and difficulty to purchase cyclodextrins, may explain why their use in pharmaceutical compounding is currently not a viable alternative.
Evaluar la utilización de ciclodextrinas como excipientes en formulación magistral desde el punto de vista galénico, biofarmacéuticofarmacocinético, toxicológico, regulatorio, económico y comercial.
MétodoBúsqueda bibliográfica de artículos de revisión y originales con alto índice de citas, consulta de documentos regulatorios y legislativos y de farmacopeas de reconocido prestigio.
ResultadosLa solubilidad, la resistencia a la hidrólisis y la eficiencia de complejación varían según la ciclodextrina y el fármaco que se pretende complejar. En algunos casos es necesario añadir excipientes para mejorar la eficiencia de complejación. Los procesos de complejación de fármacos con ciclodextrinas a nivel de laboratorio son poco robustos y además existe mucha variabilidad para una misma ciclodextrina entre proveedores y lotes de un mismo proveedor, requiriéndose más controles en las fórmulas elaboradas. La estabilidad de los complejos ciclodextrinas-fármaco puede alterar la biodisponibilidad oral de los fármacos. Además, algunas ciclodextrinas optimizan la permeabilidad a través de membranas biológicas específicas y el tiempo de contacto con las mismas. Aunque son seguras, superados determinados umbrales de dosis y tiempos de administración pueden producir efectos secundarios. Solo las ciclodextrinas que están reconocidas como excipientes en farmacopeas pueden utilizarse en formulación magistral. Las ciclodextrinas suponen un incremento del coste en formulación magistral y su adquisición a través de proveedores reconocidos no es siempre posible.
ConclusionesA pesar de sus interesantes propiedades como excipientes derivadas de la formación de complejos de inclusión, la necesidad de mayores controles de calidad, estudiar constantes de estabilidad, su alto coste y difícil adquisición pueden explicar por qué la utilización de ciclodextrinas en formulación magistral no se considera una alternativa viable en la actualidad.
Ever since its accidental discovery in 1891 by French scientist A. Villiers, who isolated it from a bacterial digest of starch, and following the subsequent identification, structural elucidation and determination in the 1940’s of its physicochemical properties, cyclodextrins (CD) have been widely used for the preparation of cosmetic, dietary and pharmaceutical products1,2.
CDs are natural cyclic oligosaccharides made up of 6 (α-cyclodextrin), 7 (β-cyclodextrin) or 8 (γ-cyclodextrin) glucose monomers connected by α-1,4-O-glycosídic bonds3. One of the main characteristics of CDs is the truncated cone shape they assume when dissolved in an aqueous solution, with an internal hydrophobic cavity and an hydrophilic outer surface that confer them an amphiphilic nature. This allows CDs to form inclusion complexes comprising highly versatile molecules, which is a highly attractive pharmaceutical feature that makes them very useful in the formulation of different kinds of medicines. Indeed, CDs are able to increase the apparent solubility and dissolution rate of poorly soluble molecules in aqueous solutions4. CDs are also able to mask unpleasant odors and flavors, reduce the irritating effect of the molecules they form complexes with, and optimize molecular diffusion through biological membranes. They may even increase the stability of several molecules in the face of multiple degradation processes such as hydrolysis, oxidation, volatilization, sublimation, heat, light, or reactions with other components present in a formulation1–4. Moreover, in view of the few incompatibilities they present with and their robust safety profile, use of CDs as pharmaceutical excipients has been approved for approximately 40 years in both Japan and Europe and for 20 years in the United States1. Although their use in orally-administered or ophthalmic formulations is particularly relevant from a compounding perspective for hospital pharmacy departments, CDs are available in many other dosage forms, with parenteral formulations constituting a better alternative than other techniques for solubilizing poorly water soluble drugs, and solid forms constituting a good alternative for improving the bioavailability of oral forms in the case of industrially produced medicines1–4. Examples of some of the commercialized medicines currently available in Spain that contain cyclodextrins as an excipient include Caverject dual® (alprostadil) solution for injection (α-cyclodextrin; α-CD), Yaz® (drospirenone/ ethynyl estradiol) tablets (β-cyclodextrin; β-CD), Nicorette® (nicotine) sublingual tablets (β-cyclodextrin, β-CD), Propulsid® (cisapride) suppositories (2-hydroxypropyl-β-cyclodextrin; HPβCD), Vfend® (voriconazole) intravenous solution (sulfobutyl ether-β-cyclodextrin sodium salt; SBEβCD) and CardioTec® (99Tc-teboroxime) solution for injection (γ-cyclodextrin; γ-CD)2.
Despite the apparent potential of CDs as excipients, they are currently seldom used in extemporaneous preparations. Multiple studies have been devoted to analyzing the possibility of using CDs to increase the apparent solubility of medicines or augment the stability of liquid medicines such as isoniazid, rifampicin or hydrochlorotiazide5,6. Some academic studies have also been published on the use of CDs as drug release modulators. For example, Shende et al. studied the use of β-CD to formulate calcium carbonate nanosponges to treat hyperphosphatemia7, and Lembo et al. showed that including acyclovir in carboxylated β-CD nanosponges increases their virucidal activity against the herpes simplex virus type 1 (HSV-1) in cell cultures8.
Taking into account that use of CDs is authorized in Spain, as well as the fact that they boast a good safety profile, it would not be unreasonable to consider adding CD to extemporaneous preparations in cases where the drug being formulated is poorly soluble or to mask its unpleasant odor or flavor. In fact, the Spanish Society of Hospital Pharmacists has promoted studies where CDs were used in extemporaneous preparations (e.g., eyedrops), involving drugs such as econazole, where a-CD was used as a drug solubilizing agent9. Voriconazole-based ophthalmic hydrogels and tacrolimus-based eyedrops have also been formulated using CDs (hydroxypropyl-β-cyclodextrin [HPβCD] or hydroxypropyl-γ-cyclodextrin [HPγCD] in the case of the former and HPβCD in the case of the latter), which have been shown to improve the solubility, permanence and ophthalmic safety of the active ingredients10,11.
The present review is aimed at evaluating the use of CDs as excipients in extemporaneous preparations from the compounding, biopharmaceutical–pharmacokinetic, toxicologic, regulatory, economic and commercial points of view.
MethodsA search was conducted in PubMed and Google Scholar of articles containing the following terms: cyclodextrins AND pharmaceutics, applications, formulation, pharmaceutical preparations, pharmaceutical compounding, drug compounding, complexation efficiency, cyclodextrin complex, inclusion complex, complexation, complex synthesis, complex formation, pediatrics, toxicology, safety and cost. Both highly cited originals and reviews were included in the study, regardless of their year of publication. The information collected from the articles selected was supplemented by an analysis of the literature references they contained and of similar papers suggested by the search engine or by the scientific journal where the articles were published. Additional sources such as books on pharmaceutical technology were consulted, as well as legislative and regulatory documents published in the Spanish Official Gazette or by the Spanish Agency for Medicines and Medical Devices and the European Medicines Agency. The literature references of each consulted document and CD monographs included in well-established pharmacopeias were also reviewed.
ResultsFollowing an exhaustive perusal of 14 review articles, 5 legislative articles, 1 book chapter and 22 original articles, it was decided that the following compounding, biopharmaceutical-pharmacokinetic, toxicologic, regulatory, economic and commercial considerations had to be analyzed before considering the use of CDs as excipients in extemporaneous formulations.
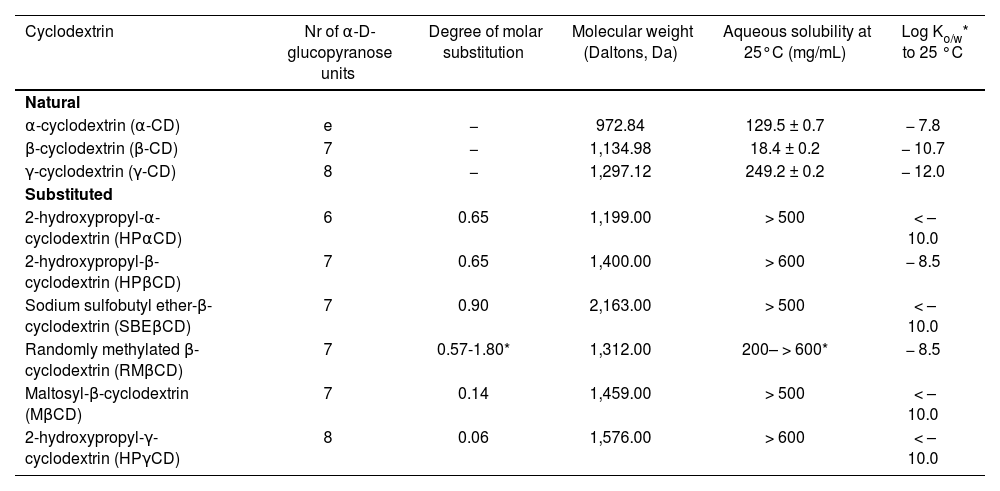
Pharmaceutical compounding considerationsFirstly, it must be pointed out that there are natural CDs and modified or substituted CDs. It is essential to understand the differences between them in terms of solubility, stability against hydrolysis and complexation efficiency, as well as the physicochemical and structural characteristics of the drugs with which CDs are meant to be complexed (Table 1)1,2.
. Characteristics of the main cyclodextrins used as excipients in pharmaceutical products: solubility, lipophilia, molecular weight and degree of molar substitution1, 2
| Cyclodextrin | Nr of α-D-glucopyranose units | Degree of molar substitution | Molecular weight (Daltons, Da) | Aqueous solubility at 25°C (mg/mL) | Log Ko/w* to 25 °C |
|---|---|---|---|---|---|
| Natural | |||||
| α-cyclodextrin (α-CD) | e | − | 972.84 | 129.5 ± 0.7 | − 7.8 |
| β-cyclodextrin (β-CD) | 7 | − | 1,134.98 | 18.4 ± 0.2 | − 10.7 |
| γ-cyclodextrin (γ-CD) | 8 | − | 1,297.12 | 249.2 ± 0.2 | − 12.0 |
| Substituted | |||||
| 2-hydroxypropyl-α-cyclodextrin (HPαCD) | 6 | 0.65 | 1,199.00 | > 500 | < – 10.0 |
| 2-hydroxypropyl-β-cyclodextrin (HPβCD) | 7 | 0.65 | 1,400.00 | > 600 | − 8.5 |
| Sodium sulfobutyl ether-β-cyclodextrin (SBEβCD) | 7 | 0.90 | 2,163.00 | > 500 | < – 10.0 |
| Randomly methylated β-cyclodextrin (RMβCD) | 7 | 0.57-1.80* | 1,312.00 | 200– > 600* | − 8.5 |
| Maltosyl-β-cyclodextrin (MβCD) | 7 | 0.14 | 1,459.00 | > 500 | < – 10.0 |
| 2-hydroxypropyl-γ-cyclodextrin (HPγCD) | 8 | 0.06 | 1,576.00 | > 600 | < – 10.0 |
Log Ko/W: oclanol/waler parlilion coefficient.
As regards solubility, natural CDs (α-CD, β-CD and γ-CD) are associated with lower water solubility than their open chain oligosaccharide counterparts. Specifically, β-CD is the least soluble of all CDs as it forms the largest amount of intermolecular hydrogen bonds and is highly prone to form intermolecular aggregates. As far as substituted CDs are concerned (HPpCD, sulfobutyl ether-β-cyclodextrin [SBEβCD], HPγCD, randomly methylated β-cyclodextrin [RMβCD], etc.), aqueous solubility is greater than that of their natural predecessors. This is because the introduction of hydroalkyl, methyl, or sulfoalkoxyl substitutes in the free hydroxyl groups of the α-D-glucopyranose units of natural CDs induces a conversion from the crystalline state in natural variants to an amorphous state in substituted variants, increasing the solubility of the latter in aqueous media1. Although it would seem that, given their higher solubility in water, substituted CDs should be the agents of choice in the formulation of aqueous solutions2, performance of further studies is recommended to determine the stability of the solutions obtained. Such stability studies are particularly important in cases where natural CDs are employed given the latter's greater tendency to form intermolecular aggregates. In the specific case of β-CD, which is the least soluble of CDs, it has been demonstrated that adding hydroxy acids such as citric acid or tartaric acid, frequently used as pH stabilizers in liquid formulations or as effervescent agents in solid formulations, may optimize aqueous solubility by means of a mechanism based on the modification of intra- and intermolecular hydrogen bonds, thereby increasing the stability of the solution1,2. Another aspect to be born in mind with respect to solubility is the temperature at which full solubilization of a given CD is achieved. In the case of α-CD, for example, the aqueous medium must be heated at around 40 °C to achieve complete dissolution and allow the formation of inclusion complexes with econazole for eyedrop formulations9. In these cases an evaluation of the effect of temperature on the stability of the solution should be carried out as part of the pharmacopeial validation of the formulation as temperature variations might alter the quality of thermolabile or volatile drugs.
Furthermore, the aggregation ability of natural CDs, particularly β-CD may in turn contribute to an increased solubilization of some drugs with which they form inclusion complexes1,2. In these cases, rather than solutions, CDs contribute to the formation of disperse, low physicochemical stability suspension-like systems. For these systems to be formed, formulations must include stabilizers such as humectants, thickeners, flocculating agents, etc., which augment the cost of the formulation by increasing the number of excipients to be used and requiring routine quality control tests of the different extemporaneous formulations prepared as part of the pharmaceutical validation process2,10. As a result, it would be justified in thinking that substitute CDs (HPpCD, SBEpCD, HP yCD, RMpCD) may be the preferred ingredients for formulating extemporaneous preparations. In fact, β-CD, given its poorer solubility and its higher aggregate formation risk, is used mainly as an excipient in solid pharmaceutical formulations’-1–4,7,8,10,12.
Hydrolysis resistanceIf CDs are to be used as excipients in orally administered pharmaceutical formulations, it is important to consider their resistance to hydrolysis, particularly in acid media. With the exception of γ-CD, which degrades rapidly in acid media, all CDs preserve their structure and their inclusion complex-forming function intact along the entire gastrointestinal tract until they reach the colon, where they are degraded by the gut bacterial flora2. γ-CD is the most appropriate CD for formulating extemporaneous preparations for parenterally administered aqueous solutions. This is due to its faster hydrolysis rate and lower nephrotoxicity risk, although it has been found that other substituted CDs such as HPpCD and RMpCD may also be used in such cases13–16.
Formation of CD-drug inclusion complexesIt is important to understand the factors intervening in the formation of CD-drug inclusion complexes.
First of all, it must be considered the molecular weight and the chemical structure of the drug to be used so as to select the most appropriate CD2. The chief requirement for forming an appropriate inclusion complex is that it should be possible to totally or partially introduce the host molecule, i.e. the drug, into the CD cavity”2,7. In the case of natural CDs, cavity sizes increase in the order of a < p < y'2. Moreover, some studies have shown that the complexation efficiency of CDs may be boosted by some of the structural characteristics of the drugs involved. This is the case of α-CD, which results in a higher inclusion efficiency in the presence of drugs based on dicarboxylic and hydrocarboxylic acids such as azelaic acid and acetylsalicylic acid that other CDs’. As a rule, it is important to take into consideration that the complexation efficiency of CDs tends to be favored by the presence of hydrophobic groups in the host molecules and hindered by the presence of polar or ionizable groups. Nonetheless, as explained in the section devoted to the strategies for improving complexation efficiency below, there are exceptional cases where this rule does not apply4,18.
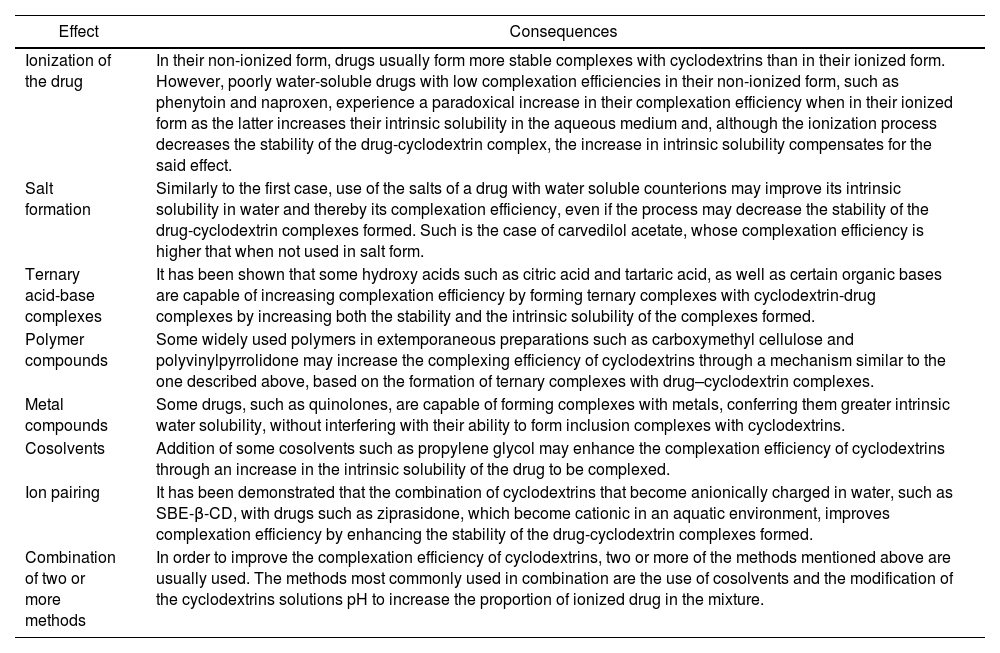
Secondly, it is important to consider the total mass of CD needed to fully complex the drug and, therefore, the total mass of the complexes formed, or bulk mass. An excessive high bulk mass may cause problems not only from the pharmaceutical but also from the toxicological point of view. Specifically, in the case of liquid orally administered or ophthalmic pharmaceutical formulations, a high bulk mass may affect the stability of the formulation given the risk of aggregate formation, especially when natural CDs are used1,2. In the case of solid dosage forms, such as tablets or capsules, a high bulk mass could cause an increase in the unit volume of the dosage form, leading to swallowing difficulties. Conversely, in the case of liquid parenterally administered formulations it is advisable to use minimal amounts of CDs as, apart from the problems related to the stability of high-bulk-mass parenteral solutions, a high amount of CD administered over a long period of time could cause renal toxicity or even hemolysis13,14. For these reasons, several optimizing strategies have been applied, such as modifying the formulations’ pH, the use of acids or bases or water soluble polymers, cosolvents, counterions or metals, which not only contribute to enhancing complexation efficiency but also the intrinsic solubility of the drug itself (Table 2)2,12. These strategies contribute to reducing the bulk mass, increasing the stability and swallowability of orally administered formulations, and reducing the toxicity risk.
Brief description of the referenced strategies used to optimize inclusion complex formation, based on the principle that complexation efficiency is directly dependent on the intrinsic solubility of the drug to be complexed and the stability constant of the cyclodextrin-drug complexes formed2, 12
| Effect | Consequences |
|---|---|
| Ionization of the drug | In their non-ionized form, drugs usually form more stable complexes with cyclodextrins than in their ionized form. However, poorly water-soluble drugs with low complexation efficiencies in their non-ionized form, such as phenytoin and naproxen, experience a paradoxical increase in their complexation efficiency when in their ionized form as the latter increases their intrinsic solubility in the aqueous medium and, although the ionization process decreases the stability of the drug-cyclodextrin complex, the increase in intrinsic solubility compensates for the said effect. |
| Salt formation | Similarly to the first case, use of the salts of a drug with water soluble counterions may improve its intrinsic solubility in water and thereby its complexation efficiency, even if the process may decrease the stability of the drug-cyclodextrin complexes formed. Such is the case of carvedilol acetate, whose complexation efficiency is higher that when not used in salt form. |
| Ternary acid-base complexes | It has been shown that some hydroxy acids such as citric acid and tartaric acid, as well as certain organic bases are capable of increasing complexation efficiency by forming ternary complexes with cyclodextrin-drug complexes by increasing both the stability and the intrinsic solubility of the complexes formed. |
| Polymer compounds | Some widely used polymers in extemporaneous preparations such as carboxymethyl cellulose and polyvinylpyrrolidone may increase the complexing efficiency of cyclodextrins through a mechanism similar to the one described above, based on the formation of ternary complexes with drug–cyclodextrin complexes. |
| Metal compounds | Some drugs, such as quinolones, are capable of forming complexes with metals, conferring them greater intrinsic water solubility, without interfering with their ability to form inclusion complexes with cyclodextrins. |
| Cosolvents | Addition of some cosolvents such as propylene glycol may enhance the complexation efficiency of cyclodextrins through an increase in the intrinsic solubility of the drug to be complexed. |
| Ion pairing | It has been demonstrated that the combination of cyclodextrins that become anionically charged in water, such as SBE-β-CD, with drugs such as ziprasidone, which become cationic in an aquatic environment, improves complexation efficiency by enhancing the stability of the drug-cyclodextrin complexes formed. |
| Combination of two or more methods | In order to improve the complexation efficiency of cyclodextrins, two or more of the methods mentioned above are usually used. The methods most commonly used in combination are the use of cosolvents and the modification of the cyclodextrins solutions pH to increase the proportion of ionized drug in the mixture. |
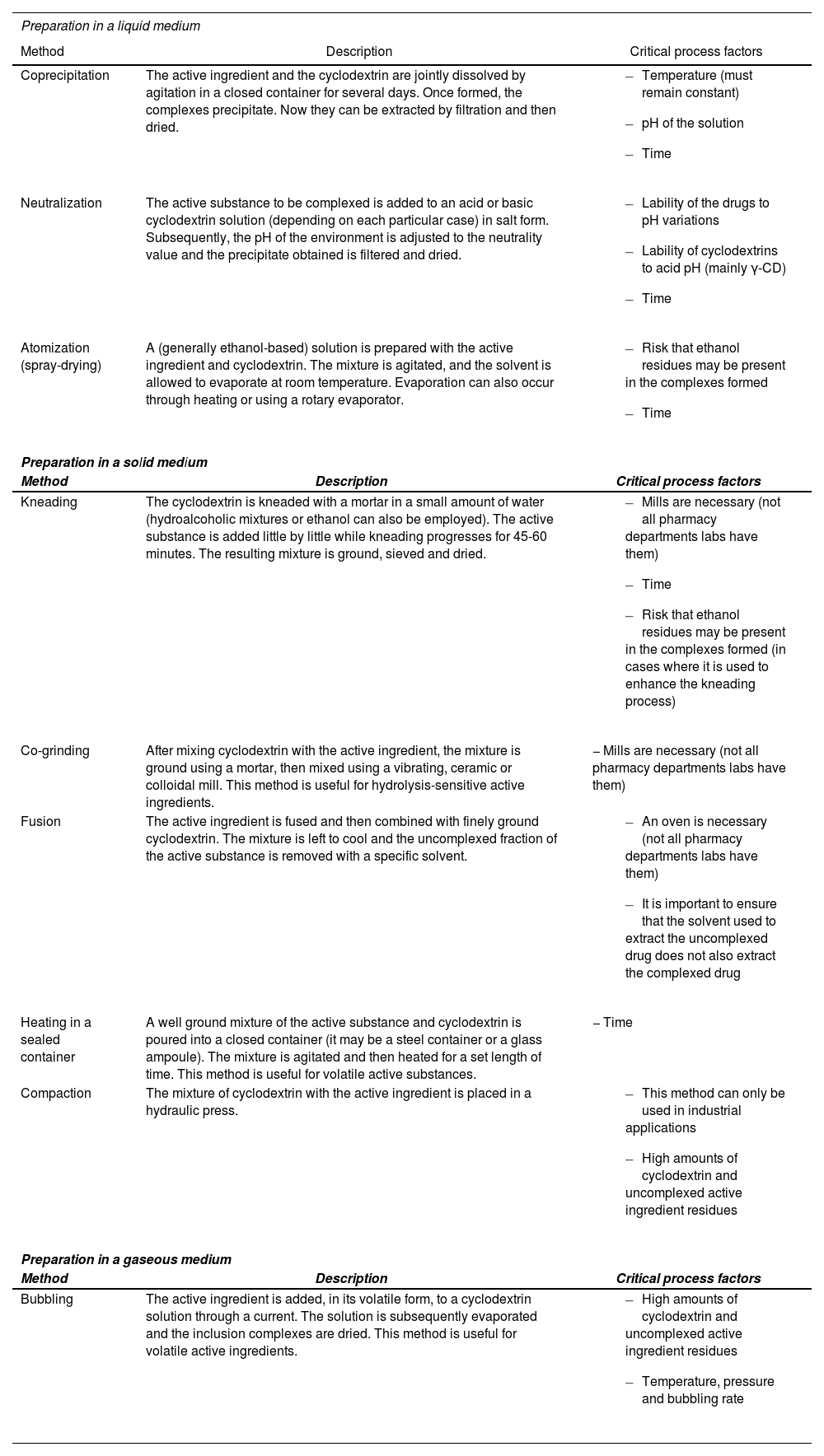
Lastly, it must be considered the difficulties inherent conducting a pharmaceutical validation of the use of cyclodextrins in extemporaneous preparations. The processes involved in preparing drug inclusion complexes are exposed to the same high level of inter-process variability as industrial drug manufacturing. This is due to the significant role played by a series of factors that may be critical for CD-drug inclusion complexes to be formed successfully at laboratory scale (Table 3)19–25. On the other hand, it has also been shown that the ability to form CD inclusion complexes is very much influenced by the environment in which CD is chemically synthesized, with complexation efficiency differences having been found not only among manufacturers but also between batches from the same manufacturer. This is particularly obvious in the case of substituted CDs given the random introduction of their substituents during the manufacturing process, both as regards their number and their position in the oligosaccharide chain1,2. In this respect, Pitha et al.26 showed that the etherification of the free hydroxyl groups at positions 2 and 3 of the β-CD ring gives rise to substituted CDs of less complexation efficiency than that observed when the etherification takes place in the free hydroxyl group at position 6. These authors also showed that a higher degree of hydroxyl etherification leads to lower complexation efficiency, most probably due to the steric impediment generated in the derivatives obtained26. In another study published a couple of years later, the same authors evaluated the influence of pH on CD etherification and concluded that, in the case of β-CD derivatives, pH differences barely influence the etherification of their hydroxyl groups, although the same conclusions cannot be drawn in the case of α-CD and γ-CD derivatives27. In view of these results, Loftsson et al. concluded in their different reviews that environmental pH should, during CD synthesis, be a constant value, and that the small pH variations taking place in the course of these reactions may give rise to derivatives with different complexation efficiencies1,2.
Methods reported in the literature to prepare inclusion complexes together with their critical process factors19, 25
| Preparation in a liquid medium | ||
|---|---|---|
| Method | Description | Critical process factors |
| Coprecipitation | The active ingredient and the cyclodextrin are jointly dissolved by agitation in a closed container for several days. Once formed, the complexes precipitate. Now they can be extracted by filtration and then dried. |
|
| Neutralization | The active substance to be complexed is added to an acid or basic cyclodextrin solution (depending on each particular case) in salt form. Subsequently, the pH of the environment is adjusted to the neutrality value and the precipitate obtained is filtered and dried. |
|
| Atomization (spray-drying) | A (generally ethanol-based) solution is prepared with the active ingredient and cyclodextrin. The mixture is agitated, and the solvent is allowed to evaporate at room temperature. Evaporation can also occur through heating or using a rotary evaporator. |
|
| Preparation in a solid medium | ||
| Method | Description | Critical process factors |
| Kneading | The cyclodextrin is kneaded with a mortar in a small amount of water (hydroalcoholic mixtures or ethanol can also be employed). The active substance is added little by little while kneading progresses for 45-60 minutes. The resulting mixture is ground, sieved and dried. |
|
| Co-grinding | After mixing cyclodextrin with the active ingredient, the mixture is ground using a mortar, then mixed using a vibrating, ceramic or colloidal mill. This method is useful for hydrolysis-sensitive active ingredients. | − Mills are necessary (not all pharmacy departments labs have them) |
| Fusion | The active ingredient is fused and then combined with finely ground cyclodextrin. The mixture is left to cool and the uncomplexed fraction of the active substance is removed with a specific solvent. |
|
| Heating in a sealed container | A well ground mixture of the active substance and cyclodextrin is poured into a closed container (it may be a steel container or a glass ampoule). The mixture is agitated and then heated for a set length of time. This method is useful for volatile active substances. | − Time |
| Compaction | The mixture of cyclodextrin with the active ingredient is placed in a hydraulic press. |
|
| Preparation in a gaseous medium | ||
| Method | Description | Critical process factors |
| Bubbling | The active ingredient is added, in its volatile form, to a cyclodextrin solution through a current. The solution is subsequently evaporated and the inclusion complexes are dried. This method is useful for volatile active ingredients. |
|
Taking into consideration the high variability of CDs, which results from their chemical synthesis process, the inconsistencies that characterize complexation processes in the laboratory setting, and the low reproducibility associated to both processes, performance of routine quality control tests is necessary following compounding, not only of each batch but also of each individual extemporaneous preparation where CDs are used as excipients.
Biopharmaceutical-pharmacokinetic considerationsConcerning the use of CDs to enhance the solubilization of orally administered drugs, it must be considered that CDs have been shown to increase the bioavailability of the FDA's biopharmaceutics classification system class II drugs, which are poorly water soluble and highly permeable. In contrast, class I (highly water soluble and highly permeable) and class III (high solubility and low permeability) drugs may reduce bioavailability1,4. This interesting aspect should be born in mind when coformulating multiple active substances in one single extemporaneous preparation.
Moreover, it must be remembered that the stability of the resulting inclusion complexes may impact bioavailability both in terms of degree and dynamics. Indeed, when the stability of such complexes is high, the release and dissolution rate of the drug may slow down, which may speed down absorption17. On the other hand, use of low-stability inclusion complexes may speed up the release of the drug in the gastrointestinal tract, leading to its precipitation in the gut. In these cases, although complexation efficiency may be satisfactory, the amount of medication absorbed may be lower, which would therefore decrease its degree of bioavailability28.
As regards the preparation of pediatric extemporaneous formulations, there is the added difficulty that the administration of many of the drugs for which CD complexation is indicated both to optimize bioavailability and to mask their unpleasant organoleptic characteristics (i.e., isavuconazole) needs to be monitored in these patients. Alterations in the dynamics or the degree of bioavailability, caused by highly stable or low-stability inclusion complexes respectively, may result in changes in the plasma levels achieved by drugs that may otherwise be regarded as appropriate29.
As far as ophthalmic administration is concerned, use of CDs may help improve not only the solubility and the stability of certain drugs but also their permeability through the tear film thereby reducing local irritation17. In the specific case of eyedrops, α-CD is considered the CD of choice as it is capable of directly interfering with the lipid membranes of the epithelial layer, increasing their porosity to the administered drug. Some authors have reported that substituted CDs such as HPβCD can also be used in these applications15,16,30,31.
When preparing a topically administered ointment or cream based on a drug that one wishes to complex in order to enhance its solubility and mask its irritating properties, it is vital to, first of all, ensure that no incompatibilities exist between CD and the other excipients. In these types of preparations, CDs are associated with a higher risk of lump and foam formation in the emulsions. If CDs are to be used in the formulation, it is best to use HPβCD as, apart from its surfactant properties, it also increases the drug-skin contact time. Other substituted CDs may also be employed15,32,33.
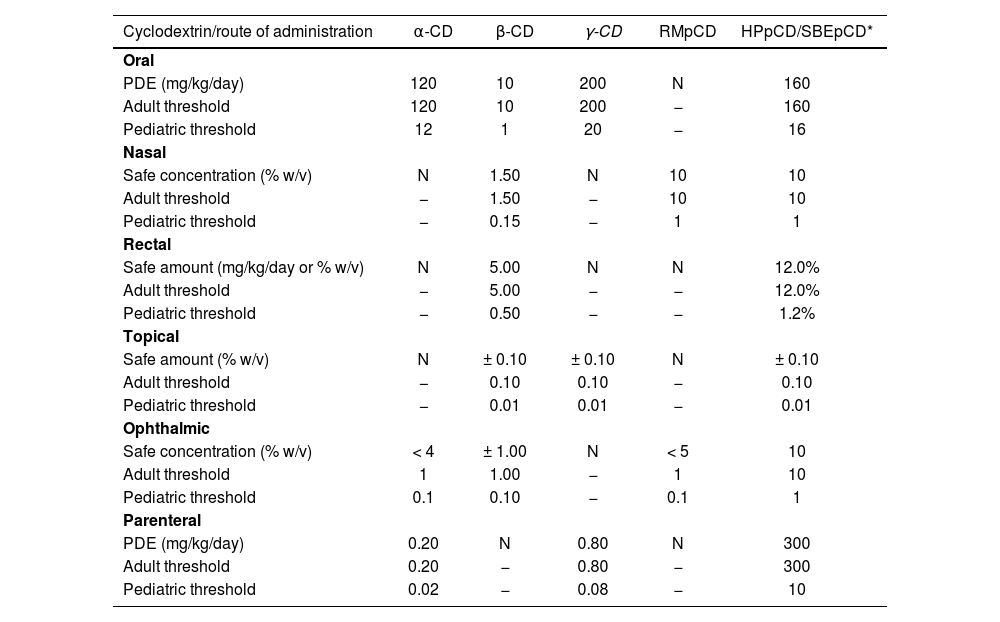
Toxicological considerationsAlthough CDs have generally been shown to be highly safe excipients, it is essential to take into consideration that administration of doses above the recommended threshold, and/or for long periods of time, may result in side effects. It is for that reason that it is important to consider not only the advisability of using a given CD in an extemporaneous preparation in terms of pharmaceutical and biopharmaceutic/pharmacokinetic criteria, but also the maximal doses established by the different European regulations for the different CDs authorized for pharmaceutical use (Table 4)15,16.
Recommended cyclodextrin dosage threshold values in the adult and pediatric population as established by the Committee for Human Medicinal Products on November 20th, 2014. Exceeding these threshold values may result in side effects associated to the use of cyclodextrin15
| Cyclodextrin/route of administration | α-CD | β-CD | γ-CD | RMpCD | HPpCD/SBEpCD* |
|---|---|---|---|---|---|
| Oral | |||||
| PDE (mg/kg/day) | 120 | 10 | 200 | N | 160 |
| Adult threshold | 120 | 10 | 200 | − | 160 |
| Pediatric threshold | 12 | 1 | 20 | − | 16 |
| Nasal | |||||
| Safe concentration (% w/v) | N | 1.50 | N | 10 | 10 |
| Adult threshold | − | 1.50 | − | 10 | 10 |
| Pediatric threshold | − | 0.15 | − | 1 | 1 |
| Rectal | |||||
| Safe amount (mg/kg/day or % w/v) | N | 5.00 | N | N | 12.0% |
| Adult threshold | − | 5.00 | − | − | 12.0% |
| Pediatric threshold | − | 0.50 | − | − | 1.2% |
| Topical | |||||
| Safe amount (% w/v) | N | ± 0.10 | ± 0.10 | N | ± 0.10 |
| Adult threshold | − | 0.10 | 0.10 | − | 0.10 |
| Pediatric threshold | − | 0.01 | 0.01 | − | 0.01 |
| Ophthalmic | |||||
| Safe concentration (% w/v) | < 4 | ± 1.00 | N | < 5 | 10 |
| Adult threshold | 1 | 1.00 | − | 1 | 10 |
| Pediatric threshold | 0.1 | 0.10 | − | 0.1 | 1 |
| Parenteral | |||||
| PDE (mg/kg/day) | 0.20 | N | 0.80 | N | 300 |
| Adult threshold | 0.20 | − | 0.80 | − | 300 |
| Pediatric threshold | 0.02 | − | 0.08 | − | 10 |
N: no reference dala is available for the corresponding route of administration; PDE: permitted daily exposure. ±1 = estimation based on the properties of cycledextrine.
As regards oral CD administration, it has been shown that only insignificant amounts of CD are absorbed during colonic transit by intestinal bacteria, RMβCD showing the highest absorption rates, with an oral bioavailability rate of 12% in rats, as compared with 0.1-3% for the other CDs. There is, however, evidence from animal studies that high doses of orally administered CDs (> 1,000 mg/kg/day) may result in reversible diarrhea and cecal enlargement2,15,16,34–36.
It has also been shown that the presence of specific components in the formulation or a given concentration of the CD used may enhance absorption of CDs when administered by other routes of administration. Such is the case of the intranasal and the intratracheal routes, for which systemic absorption of CDs is practically negligible. Specifically, administration of drugs containing very high doses of CDs could have an impact on the absorption not only of the complexed drug but also of the CDs themselves13,29–31,37. It has been shown that, following intranasal administration of dipropyl-β-cyclodextrin (not used in pharmaceutical products) at a concentration of 80 mM in rats, as much as 16% of the dose is excreted in the urine. Moreover, in the case of HPβCD it has been shown that the presence of absorption-stimulating agents increases absorption of CD, with urine excretion rates of 45% of the administered dose, as compared with only 3% when HPβCD is administered on its own15,34. Likewise, topical absorption of CDs is practically nonexistent, although when CD is combined with absorption stimulators such as HPE-101 or with occlusive excipients, absorption rates of 12% have been found for β-CD, 43% for RMβCD and 53% for HPβCD, albeit without evidence of any adverse events15,34–36.
The rectal route, for its part, has shown that absorption of CDs may be significant as they may act as drug absorption optimizers and be absorbed accordingly17. Nevertheless, the risk of toxic reactions following rectal absorption is not as high as in the case of parenteral administration, where high doses of CDs may cause severe toxicity at renal and hematological level13–16,34. Intravenous administration of CD-containing pharmaceutical preparations may induce changes in cell morphology as well as hemolysis, possibly due to the complexation of cell membrane components such as cholesterol, phospholipids and proteins. Such hemolytic activity seems to follow the β- > α- > γ-cyclodextrin order38.
From a safety standpoint, the most appropriate natural CD for parenteral administration is γ-CD, which is not associated with significant nephrotoxicity (probably as a result of its rapid enzymatic degradation) and presents with low hemolytic activity. There are however some β-CD derivatives that may be safely used in parenteral administration such as HPβCD and SBEβCD, provided that they are administered in amounts above 15 mg/kg/day and that the length of administration of the extemporaneous preparation containing the CD in question is not too prolonged13–16.
Regulatory considerationsIt is important to bear in mind that not all the available CDs may be used in extemporaneous preparations, regardless of their favorable properties and their good safety profile. As established in Royal Decree 175/2001 of 23 February 2001, which sets out the procedures to be followed for compounding and controlling the quality of extemporaneous preparations and officinal formulations, all the raw materials used in extemporaneous preparations must be warranted by a specific monograph in the Royal Spanish Pharmacopeia or, failing that, in the European Pharmacopeia or in some other well-established pharmacopeia39. The Royal Spanish Pharmacopeia currently recognizes natural CDs (α-CD and β-CD) and HPβCD as pharmaceutical grade excipients. Both supplement 8.7 of the European Pharmacopeia and the 2017 edition of the British Pharmacopoeia include monographs for γ-CD and dimethyl-β-cyclodextrin (DMβCD), which is nowadays seldom used. The American Pharmacopeia (USP-40) only recognizes γ-CD as an excipient, although the FDA's inactive ingredients list also includes HPβCD and SBEβCD2. In view of the above, it is not advisable to use CDs not included in any of the mentioned pharmacopeias, even if their use may be referenced in the literature.
Furthermore, it should be mentioned that natural CDs (α-CD, β-CD and γ-CD) are included in the FDA's GRAS List as flavor stabilizers, which means they could be a valid alternative for masking the unpleasant taste of pediatric pharmaceutical formulations2. However it must be remembered that, as their water solubility is lower, they should be formulated as suspensions in these applications12. As mentioned above, when used in solution form, the formulations should be subject to stability studies.
Economic and commercial considerationsAlthough their price has declined in the recent past, CDs are still expensive excipients. The relatively high costs associated to the use of CDs in extemporaneous preparations, combined with the need to perform frequent quality control tests as a result of the variability across different manufacturers, and even batches from the same manufacturer, for one same cyclodextrin, and the exhaustive pharmaceutical validations that must be carried out of the formulations prepared, require an increased time investment by the compounding laboratory staff. If we add to this the fact that it is at times necessary to include some extra excipient to the extemporaneous preparation in order to boost its complexation efficiency, it is clear that the cost of the formulation may experience a substantial increase40.
On the other hand, the impossibility to access these components through the usual vendors included in the Spanish Unified Registry of Active Substance Providers (RUESA) constitutes a significant impediment to acquiring these raw materials. The Registry only includes HPβCD, added in 2021. Its indications are parentally-administered drugs and treatment of Niemann-Pick disease, for which it has been designated an “orphan drug”41. This means that if a decision was made to incorporate a different cyclodextrin to a routinely-formulated extemporaneous preparation it would be necessary to resort to non-RUESA approved vendors, which would involve a preliminary risk analysis of the CD synthesis procedure implemented by such vendors and an evaluation of the quality assurance systems they apply, in accordance with Guidelines 2015/C 95/02 of 19th March 2015 on the formal risk evaluation required to ensure the appropriateness of the practices followed to manufacture excipients employed in medicines for human use42.
DiscussionSolubility, resistance to hydrolysis and complexation efficiency are the main factors that determine the selection of a given CD as an excipient for extemporaneous preparations. However, the inconsistency of complexation processes involving CDs at laboratory scale and the complexation efficiency variabilities across different vendors and across batches from the same vendor are the main features limiting the use of CDs from a compounding perspective, as they require the implementation of routine quality control tests for each formulation prepared.
From the biopharmaceutical-pharmacokinetic point of view, it must be remembered that the stability of CD-drug complexes may alter the medicines oral bioavailability, which makes it advisable to perform regular checks of their stability constants. In addition, some CDs optimize permeability through specific biological membranes as well as by the length of time they stay in contact with them, which is considered an important selection criterion (e.g., α-CD for the ophthalmic route, HPβCD for the topical route).
In spite of their satisfactory toxicologic profile, CDs could result in side effects beyond certain dosage thresholds and administration times.
At a regulatory level, only CDs recognized as excipients by well-established pharmacopeias may be used in extemporaneous preparations, even if the scientific evidence warrants the use of other CDs.
Lastly, considering the high cost of CDs and the fact that their use requires more resources, quality control measures and, occasionally, other excipients, it can be concluded that the use of CDs as excipients in extemporaneous preparations results in a significantly heavier workload for the compounding laboratories of hospital pharmacy departments. To the factors above should be added the difficulty of acquiring CDs through RUESAaccredited vendors.
The need to conduct stringent quality control measures for each extemporaneous preparation formulated and perform exhaustive studies of the stability constants of inclusion complexes to guarantee proper bioavailability, together with the high cost of CDs, the higher consumption of resources involved in their use and the difficulties involved in acquiring them through the usual channels, may explain why their use as excipients in extemporaneous preparations is not nowadays considered a feasible alternative despite the significant advantages inherent in their use.
FundingNo funding.
Conflict of interestNo conflicts of interests.
Early Access date (11/06/2021).

